系列二亚胺羰基Re配合物光谱的密度泛函研究*
2022-10-22慈成刚臧杰超
慈成刚,臧杰超
(黔南民族师范学院,贵州省计算催化化学重点实验室,贵州 都匀 558000)
新型有机发光二极管(OLED)材料的研发一直是化学和材料领域的重要挑战之一[1-2]。紫外-可见光谱和磷光发射光谱在OLED性能评价中扮演了重要角色,在分子层面对其研究有助于调控其发光性能。前人前期研究发现,具有d6电子组态的过渡金属元素在光催化,电催化和OLED领域显示了优异的性能[3-5]。其中,二亚胺羰基铼配合物作为典型的代表,显示出了优秀的三重态发射性能,成为重要的OLED材料和CO2光还原催化剂之一。其中,Firn等[6]合成了一组典型的二亚胺羰基铼配合物[Re(CO)3Cl(Cl2phen)],(简写为Re1)和[Re(CO)3(py)(Cl2phen)]+,(简写为Re2)(图1),并测定了二者的红外光谱,紫外-可见光谱和磷光发射光谱。但因受限于电子的快速跃迁,无法探测其前线分子轨道,发光机理和分子振动模式。而这些信息对深入研究其OLED性能和CO2光催化还原性能都具有十分重要的意义。本文采用密度泛函理论(DFT),含时密度泛函理论(TDDFT)方法对Re1和Re2的几何结构,前线分子轨道,紫外-可见光谱,磷光发射光谱和红外光谱进行模拟,给出其电子跃迁机理和分子振动模式,并对取代基吡啶和Cl-对上述性质的影响进行讨论。在此基础上,通过重组能的计算,给出Re1和Re2的OLED性能评价。这些计算将对未来系列二亚胺羰基铼配合物的实验合成,OLED性能和CO2光催化还原性能调控给出可靠的重要信息。

图1 计算的[Re(CO)3Cl(Cl2phen)],(Re1)和 [Re(CO)3(py)(Cl2phen)]+(Re2)结构Fig.1 Calculated structure of Re1 and Re2
1 计算方法
本文所有计算均采用Gaussian 16 C.01程序包[7]。几何结构优化采用PBE0杂化泛函[8-9]。对Re原子采用SDDALL基组,对C,H,N,O和Cl等非金属原子采用6-311G**基组[10]。对所有优化的结构都进行频率计算,用来确定极小能量结构。所有计算均采用严格的收敛标准(10-12au),并均采用IEF-PCM溶剂化模型,乙腈为溶剂[11]。采用GaussSum 3.0软件绘制紫外光谱。采用GaussView6.1.1软件绘制红外光谱。
2 结果和讨论
2.1 催化剂结构和前线分子轨道
采用密度泛函理论(DFT)方法,在乙腈溶剂中对Re1和Re2的几何结构进行全优化,结果列于表1所示。可以看出在优化的Re1和Re2中,Re-C(O)键平均距离为1.93 Å,Re-Cl键和Re-N(吡啶)键距离分别为2.52 Å和2.24 Å。相邻轴向的C=O基团之间的键角在88.8°~92.6°之间,表明C=O基团和Re之间呈现线性配位。因此,Re金属中心显示出一种六配位的扭曲八面体结构。因为实验上未提供XRD的结构数据,我们采用对比其他结构相似的二亚胺羰基铼配合物的XRD数据[12]。结果显示计算的Re1和Re2的结构数据和实验值很接近。

表1 PBE0泛函计算的Re1和Re2的几何结构Table 1 Selected bond lengths and angles for species Re1 and Re2 by means of PBE0 functional
为了更好的理解其OLED性能和催化活性起源,基于优化的几何结构,计算了Re1和Re2的前线分子轨道(FMO)。并绘制于图2。

图2 [Re(CO)3Cl(Cl2phen)](Re1)和 [Re(CO)3(py)(Cl2phen)]+(Re2)的前线分子轨道Fig.2 Frontier molecular orbital(FMO) of Re1 and Re2
图2显示Re1和Re2的最低空轨道(LUMO)主要来自于二亚胺配体的π*反键轨道,而最高占据轨道(HOMO)均主要来自金属Re的3d轨道和C=O基团的2p轨道的贡献,并显示出明显的σ*(Re-Cl)和σ*(Re-N)反键轨道特征。可以预测,在紫外-可见光的激发下,金属Re的电子被激发到二亚胺配体,形成空穴,诱导外界电子进入该σ*反键轨道,这将有利于配体Cl-或吡啶的解离,形成催化反应活性中心,推动后续的CO2配位和还原反应。
2.2 紫外-可见光谱和电子跃迁类型
根据Frank-Condon原理,基于PBE0泛函优化的基态几何结构,利用含时密度泛函理论(TDDFT)方法,在乙腈溶剂中,对前30个电子激发态进行计算,并将得到紫外-可见光谱和跃迁特征绘制于图3。从图3可以看出,PBE0泛函模拟的紫外-可见光谱与实验数据符合很好,这也证实了本文计算方法的可靠性。模拟的Re1紫外-可见光谱主要显示出3个最大吸收峰,分别位于228 nm,257 nm和380 nm,与实验值213 nm,269 nm和384 nm符合很好。其跃迁类型分别对应HOMO-8 → LUMO+1(228 nm),HOMO-6 → LUMO(257 nm)和HOMO-1 → LUMO+1(380 nm)。其中,LUMO和LUMO+1的主要贡献均来自二亚胺配体的π*轨道,而HOMO-8主要为二亚胺配体的π*轨道和二亚胺取代基Cl的2p轨道。因此,228 nm的吸收峰主要来自于配体L到配体L电荷转移跃迁,即LLCT。类似的,257 nm的吸收峰来自于LLCT和金属配体X到二亚胺配体L的电荷转移跃迁(XLCT),380 nm的吸收峰主要来自金属M到配体L的电荷转移跃迁(MLCT)。对于Re2,计算的紫外-可见吸收峰主要有5个,分别位于232 nm,240 nm,252 nm,262 nm和349 nm与实验值213 nm,241 nm,259 nm,278 nm和334 nm符合很好。跃迁类型分别对应LLCT和XLCT(232 nm),MXCT和MMCT(240 nm),MLCT(252 nm),LLCT和XLCT(262 nm),MLCT(349 nm)。
注意,以Re1为例,一旦电荷由Re中心基团进入二亚胺配体后,将形成类似电荷分离的{[Re(CO)3Cl]+(Cl2phen)e}特征的活性结构。前面提到,金属Re中心基团具有明显的Re-Cl键的σ*反键轨道(Re1的HOMO,图2),当电子被激发后,进入该σ*反键轨道,将有利于配体解离,形成活性中间体,推动接下来可能的催化还原反应进行,如CO2还原。再者,对比Re1和Re2,二者的唯一区别为Re中心配体分别为Cl-和吡啶。当Cl-被吡啶取代后,导致Re中心电子密度明显降低(Mulliken电荷由+0.58 e增加到+0.80 e)。这导致最大紫外-可见吸收峰由可见区向紫外区移动(Re1的380 nm到Re2的349 nm),即吸电子取代基降低了Re中心电子密度,将导致吸收峰向紫外区移动。反之,供电子取代基将提高Re中心配体的电子密度,将使最大吸收峰向可见光区移动,增加了可见光的利用,这为研发可见光驱动的OLED材料和光催化剂研发提供了理论指导。

图3 PBE0泛函计算的紫外光谱和垂直激发能Fig.3 Calculated UV spectra and vertical excited energies by means of PBE0 functional
2.3 磷光发射和电子跃迁类型
磷光发射光谱在OLED性能评价指标中扮演了重要的角色。其中,从三重态辐射跃迁回基态的过程,能够使发光量子产率得到提升。此处,基于优化的S0基态结构,对第一三重激发态(T1)结构在乙腈溶剂中进行全优化。在优化的T1结构基础上,采用TDDFT方法对磷光发射过程进行模拟。计算的Re1和Re2磷光发射峰分别位于627 nm和576 nm,与实验值600 nm和567 nm符合较好。为了进一步确认其跃迁类型,我们基于优化的T1态结构,在乙腈溶剂中,对这两个发射峰的自然跃迁轨道(NTO)进行计算,并将计算结果列于表2所示。可以看出Re1的发射光谱具有MLCT特征,而Re2的发射光谱除了MLCT跃迁外,还显示出部分的LLCT跃迁特征。再者,对比Re1和Re2,金属中心取代基由Cl-到吡啶,导致最大发射峰发生蓝移。原因与上述吸收光谱类似,即Re中心电子密度降低,导致电子跃迁需要更多的能量,使发射峰蓝移。
2.4 红外光谱和分子振动模式
红外光谱作为常规实验表征的重要手段之一,能够快速、准确的通过红外特征峰的强度、形状和频率来推测分子结构,成为化学和材料领域的重要研究手段。采用PBE0泛函,在乙腈溶剂中,对Re1和Re2结构进行全优化,在相同计算水平上计算了振动频率,并采用校正因子0.9594[13],进行频率校正。计算结果显示所有振动频率没有虚频,均为势能面上的能量极小点。采用GaussianView 6.1.1对红外光谱进行绘制。

表2 PBE0泛函计算的自然跃迁轨道(NTO)Table 2 Calculated Natural transition orbitals(NTOs) by means of PBE0 functional
PBE0泛函计算的红外光谱特征峰,分子振动模式,包括实验数据分别绘制于图4和表3。计算结果和实验测定数据符合很好,最大误差仅为8 cm-1(0.05 kcal/mol)。因为Re1和Re2的结构相似,导致二者的红外振动特征峰也很相似,其中Re1的2个吸收峰分别位于1901 cm-1和2026 cm-1,Re2的2个吸收峰分别位于1926 cm-1和2038 cm-1,均对应C=O伸缩振动。值得注意的是吡啶取代基的出现,导致Re中心正电荷密度增加,进而通过诱导效应使C=O基团中O原子的电子向C原子发生部分转移,增强了C=O键,提升了C=O的振动频率,导致Re2的特征峰相比Re1向高波数移动(Re2相比Re1增加了约12~16 cm-1)。
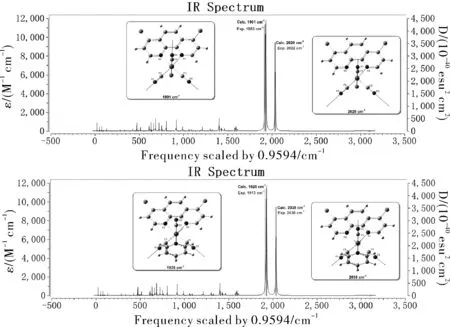
图4 计算的红外光谱和振动模式Fig.4 Calculated IR spectra and vibrational mods

表3 Re1和Re2的主要红外特征峰,振动模式和摩尔吸收系数εTable 3 Calculated infrared peaks, major vibrational modes and molar absorption coefficient, ε of Re1 and Re2
2.5 重组能
在光谱分析的基础上,为了对Re1和Re2的OLED性能进行评价,需要对OLED的发光原理进行讨论。在外界电场作用下,OLED会通过阴极产生电子,通过阳极产生空穴,注入到OLED的电子和空穴传输层,并迁移到发光层相遇,最终通过能量激子激发分子,形成可见光。可以看出电子和空穴的平衡作为重要指标,在OLED材料性能评价中扮演了重要的角色。基于优化的基态结构,分别对垂直电离势(IPv),绝热电离势(IPa),垂直电子亲和势(EAv),绝热电子亲和势(EAa),空穴抽取势(HEP)和电子抽取势(EEP)进行计算,并将计算结果列于表4所示。可以看出Re2的IP值相对Re1低约0.50 eV,即Re2具有相对较强的空穴注入能力。而Re1的EA值相比Re2高约0.20 eV,即Re1的电子注入较Re2容易。然而,Re2的高IP值和低EA值,暗示了其电荷传输的不平衡。再者,Re1和Re2的HEP值均高于EEP值,而且Re1的HEP和EEP值均高于Re2,表明Re1的绝热空穴转移和绝热电子转移均优于Re2。

表4 Re1和Re2的电离能,电子亲和势和重组能Table 4 Ionization potentials(IPs), electron affinities(EAs), and reorganization energies for complexes Re1 and Re2(eV)
此外,基于Marcus理论[14],利用计算的IP,EA,HEP和EEP,根据下列公式,对重组能(λ)进行计算:
式中,k是电荷转移速率,A是常数,kb是玻尔兹曼常数,λ是重组能,T是温度。基于前人前期工作,在固体材料中,分子间电荷传输受限,重组能成为评价电荷传输速率的重要指标。重组能越小,电荷传输越有利。其中,空穴和电子的重组能分别为:
λhole= IPv- HEP
λelectron= EEP - EAv
表4显示Re1和Re2的λhole均大于λelectron,表明二者的电子传输速率相对较好,可以作为电子传输材料的重要母体。再者Re1的λhole和λelectron差值小于Re2,显示了相对较好的电荷转移平衡能力,更适合于作为OLED的发光材料。
3 结 论
采用DFT和TDDFT方法,在乙腈溶剂中,对[Re(CO)3Cl(Cl2phen)](Re1)和[Re(CO)3(py)(Cl2phen)]+(Re2)的S0基态和T1态结构,前线分子轨道,紫外-可见光谱,磷光发射光谱,红外光谱和重组能进行计算。结果显示:
(1)PBE0泛函,结合SDDALL+6-311+G**方法计算的S0基态和T1态几何结构,紫外-可见光谱,磷光发射光谱和红外光谱与实验值符合很好。
(2)Re1和Re2的HOMO轨道均为Re中心基团的σ*反键轨道,一旦电子进入该轨道,将有利于配体Cl-或吡啶的解离和活性中间体产生,有利于后续催化反应。
(3)Re1和Re2的紫外吸收峰的显示出多种跃迁类型,即LLCT,MLCT和XLCT等。取代基导致的Re中心配体电子密度降低,使最大吸收峰蓝移。
(4)Re1和Re2的红外特征峰均为Re中心C=O键的伸缩振动。Re中心配体电子密度的降低,使Re2的振动峰高于Re1。
(5)重组能计算结果显示相比Re2,Re1更适合作为OLED发光材料。
综上,通过对Re1和Re2光谱和重组能的计算研究,将为后续实验合成相关OLED材料或CO2光还原催化剂提供帮助。
