肿瘤相关巨噬细胞靶向治疗研究进展
2022-10-19刘洋汉陈振锋
刘 慧, 李 丽, 刘洋汉, 陈振锋*
(1.省部共建药用资源化学与药物分子工程国家重点实验室 (广西师范大学), 广西 桂林 541004;2.广西民族药协同创新中心 (广西师范大学), 广西 桂林 541004; 3.广西师范大学 化学与药学学院, 广西 桂林 541004)
巨噬细胞是维持机体内环境稳定和抵御外来病原体的重要先天免疫细胞群,具有高度的可塑性,对不同的组织分区具有不同的功能。肿瘤微环境(tumor microenvironment, TME)中的肿瘤相关巨噬细胞(tumor-associated macrophages, TAMs)已被证明可以促进肿瘤细胞的增殖、免疫抑制、血管生成,以及支持肿瘤侵袭和转移。肿瘤组织中TAMs的密度往往与患者预后不良相关。随着人们对TAMs与恶性肿瘤关系研究的日益深入,TAMs被认为是肿瘤治疗的潜在靶点。本文对TAMs的来源、分类、TAMs与肿瘤的关系及靶向TAMs的治疗策略等进行综述。
1 巨噬细胞来源及其分类
巨噬细胞分布于哺乳动物全身的各个组织中,执行特定的功能。起初的证据认为,巨噬细胞的主要来源是血液中的单核细胞,这些单核细胞受组织微环境的影响,经外渗进入组织,然后进一步分化为组织驻留巨噬细胞[1-2]。然而,另有证据对这一观点提出质疑,认为组织驻留的单核巨噬细胞也可能来自卵黄囊、胎肝以及造血干细胞。如小胶质细胞来源于卵黄囊[3],皮肤、脾脏、胰腺、肝脏和腹膜腔中的主要组织驻留巨噬细胞来源于卵黄囊祖细胞或胎肝,肾、肺中的巨噬细胞可能来源于造血干细胞和卵黄囊前体细胞,而胃肠道中的巨噬细胞则来自血液中的单核细胞[4]。
一般认为,巨噬细胞有2种极化状态:经典激活(M1)巨噬细胞亚群和交替激活(M2)巨噬细胞亚群。M1型由辅助性T细胞1(Th1)型细胞因子,如干扰素-γ、细菌脂多糖和Toll样受体激动剂驱动[5]。M2型在不同的刺激下可分化为M2a、M2b、M2c和M2d型巨噬细胞[5-7](图1)。M2a巨噬细胞由Th2细胞因子IL-4或IL-13诱导;M2b巨噬细胞由免疫复合物(ICs)、LPS、TLRs或IL-1R拮抗剂诱导;M2c巨噬细胞由IL-10、转化生长因子-β(TGF-β)或糖皮质激素诱导;M2d巨噬细胞由IL-6、白血病抑制因子LIF和腺苷诱导,且表达高水平的VEGF和IL-10[5,8](表1)。然而,人类癌症中的巨噬细胞并不能简单分类为经典激活的M1型巨噬细胞或交替激活的M2型巨噬细胞,当前的M1与M2极化模型可以扩展到具有至少9个不同巨噬细胞激活程序的“谱模型”[9]。巨噬细胞表型的动态改变发生在肿瘤发生、进展和转移过程中,不同的TAMs亚群负责不同的肿瘤活动[10-12]。
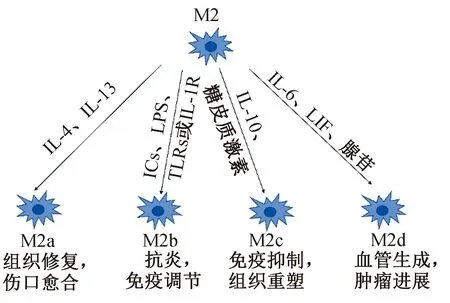
图1 M2巨噬细胞激活因子及功能[7]Fig.1 Activated factors and functions of M2 macrophages[7]
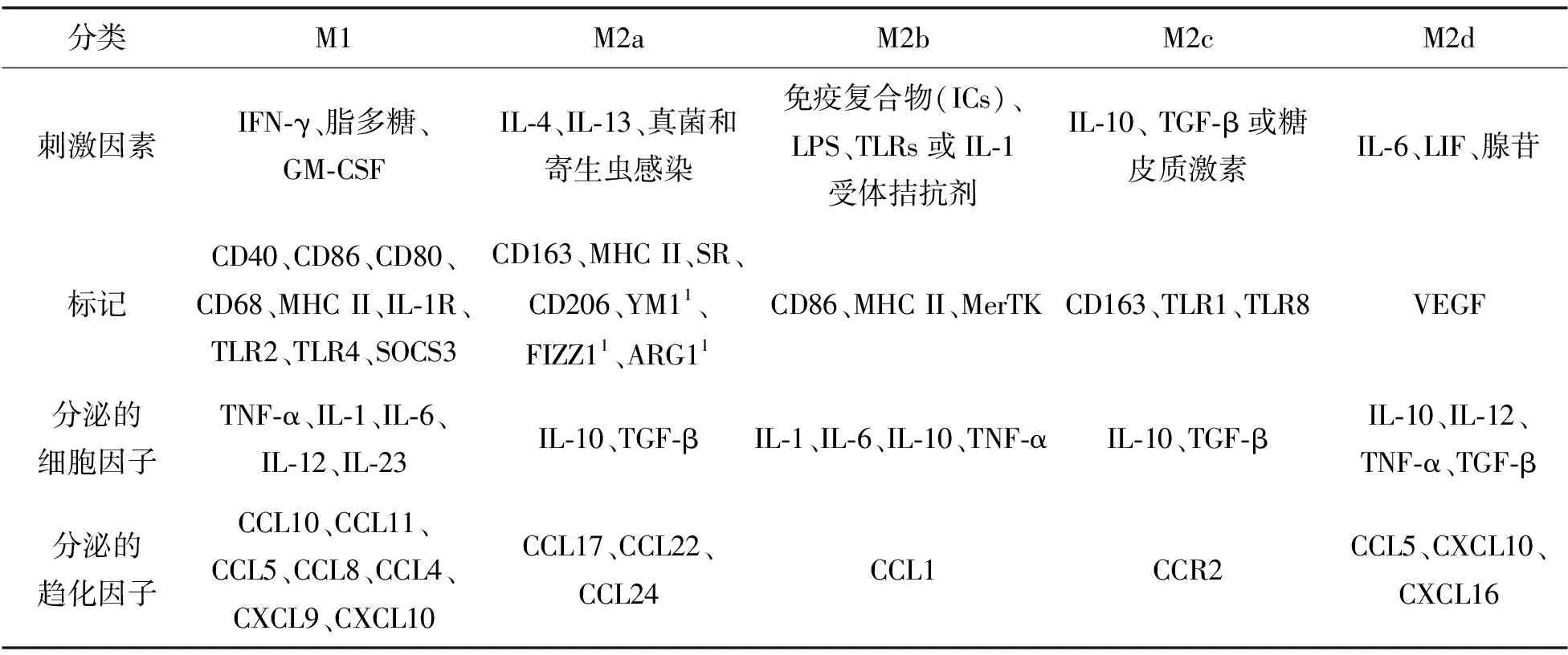
表1 经典激活的M1和交替激活的M2内型的特征[8]
1. 表示标记物仅在小鼠巨噬细胞中发现。
2 肿瘤相关巨噬细胞在肿瘤生长进程中的作用
TME由多种细胞类型(内皮细胞、成纤维细胞、免疫细胞等)和细胞外成分(细胞因子、生长因子、激素、细胞外基质等)组成,围绕在肿瘤细胞周围,由血管网络滋养[13]。作为TME中的重要成分之一,TAMs在肿瘤生长、进展的各个阶段发挥重要作用。
2.1 侵袭和转移
肿瘤细胞常常离开原发部位而形成转移集落。肿瘤细胞的远处转移能力取决于肿瘤微环境,TAMs作为肿瘤微环境的主要成分,在肿瘤转移中起着重要作用。TAMs主要通过分泌组织蛋白酶、基质金属蛋白酶(MMPs)和丝氨酸蛋白酶来促进肿瘤细胞的侵袭和迁移,这些物质可以改变细胞间的连接,并破坏基底膜。例如,TAMs来源的组织蛋白酶B可以促进乳腺癌细胞侵袭和肺转移[14]。TAMs可以诱导局部和全身水平的MMP9、几丁质酶-3样蛋白1(CHI3L1)、VEGF和脂钙素-2(Lcn-2)来介导三阴性乳腺癌小鼠模型中的肿瘤转移[15]。从98例原发性肺癌组织中分离出的TAMs高水平表达HGF、环氧合酶-2(COX-2)、VEGF-A、组织蛋白酶K、PDGF-B、MMP9和尿激酶型纤溶酶激活物(uPA),TAMs条件培养液可显著促进人各种肿瘤细胞的迁移和侵袭,而阻断uPA和MMP9则可抑制TAMs诱导的侵袭[16]。通过在体内直接观察巨噬细胞及其与肿瘤细胞的相互作用,发现几乎所有肿瘤细胞的迁移和血管内渗都与巨噬细胞有关[17]。TAMs通过分泌细胞外基质(ECM)重塑蛋白支持肿瘤细胞的黏附和侵袭[18]。上皮-间充质转化(epithelial-mesenchymal transition, EMT)在肿瘤进展和转移中起重要作用,它是TAMs与肿瘤细胞相互作用的重要结果[19]。
2.2 血管生成
1971年,Folkman等[20]提出肿瘤的生长依赖于血管生成,血管生成对于提供营养和氧气、清除废物以及癌细胞的转移都是必不可少的。如果没有血管生成,肿瘤细胞就无法生长到大于1~2 mm3,并会因缺氧而死亡。组织中存在内源性的血管生成激活剂和抑制剂,称为“血管生成开关”,在正常情况下,这种开关处于平衡状态。当这个开关“打开”时(如在肿瘤进展过程中),血管生成激活因子表达上调,导致血管生成[21]。肿瘤的缺氧或坏死区通过释放低氧诱导的趋化因子,如血管内皮生长因子、内皮素和EMAPII(也称为SCYE1)来吸引TAMs[22]。TAM表达许多血管生成调节因子,如VEGF、uPA、TNF-α、IL-1β、CXCL8、PDGF-β、MMP7、MMP9和MMP12等来促进肿瘤血管生成[23]。血管生成素2(ANG2)是血管生成素家族的成员,主要通过血管生成素受体2(TIE2)传递信号,其不仅为抗VEGF治疗提供逃逸机制,还调节TIE2表达的促血管生成TAMs的活性。在携带原位乳腺肿瘤的小鼠中,ANG2阻断抑制了肿瘤的血管生成、生长和转移,并削弱了促血管生成的TIE2+巨噬细胞的活性[24]。阻断ANG2也抑制了容易对抗VEGF/VEGFR治疗产生耐药性的小鼠模型的血管生成和肿瘤生长。同样,ANG2和VEGF的双重抑制可以在一定程度上改变TAMs极化,使小鼠胶质母细胞瘤模型的肿瘤血管正常化,延长生存期[25]。此外,一些转录因子和相关的信号转导通路也参与促血管生成过程,如STAT3通路通过促进TAMs、内皮细胞(ECs)和肿瘤细胞之间的多向串扰参与血管生成[26],TAMs分泌的WNT7b可刺激内皮细胞Wnt/β-catenin途径,促进血管生成活性[27]。
2.3 免疫抑制
TAMs还可以通过免疫抑制来促进肿瘤进展。TAMs是肿瘤中主要的免疫调节细胞,参与抑制肿瘤微环境中的细胞毒性T淋巴细胞(CTL)反应。有效的抗肿瘤免疫主要依赖于CD8+T细胞的激活。然而,TAMs采用各种机制使CTL直接或间接地失活,有助于肿瘤免疫逃逸和肿瘤发展[28]。一方面,TAMs分泌的多种趋化因子(如CCL2、CCL5、CCL17等)、细胞因子(如HGF、PDGF-B、VEGF、IL-4、IL-10等)以及酶(如组织蛋白酶K、环氧合酶-2、精氨酸酶1和基质金属蛋白酶)可直接抑制CD8+T和CD4+T细胞的效应功能。此外,来自TAMs的这些趋化因子、细胞因子和酶还可以刺激适应性调节性T细胞(ITregs)的产生,并招募天然调节性T细胞Tregs(NTregs),通过直接抑制效应性T细胞或分泌免疫抑制因子来发挥免疫抑制功能[29]。另一方面,免疫检查点分子的表达抑制T细胞免疫被认为是TAMs介导的另一种重要免疫抑制机制。如从PD-L1高表达的肝细胞癌中分离出来的TAMs可以抑制肿瘤特异性T细胞免疫,并促进肿瘤生长[30]。除了直接作用于T细胞外,TAMs还与其他免疫细胞和基质细胞协同作用,构建免疫抑制的TME。Tregs和MDSCs是肿瘤微环境中的另外2种重要的免疫抑制细胞类型,胶质瘤中的TAMs可以通过产生丰富的CCL2来招募CCR2+Ly-6C+MDSCs和CCR4+Treg细胞[31]。
2.4 化疗或放疗耐药
TAMs在化疗药物治疗中具有两重作用:有时会提高治疗效果,但更多的是导致化疗耐药。在TAMs对化疗的积极作用中,早期研究指出,巨噬细胞发挥宿主防御机制有助于阿霉素的治疗效果,这一概念已被Zitvogel课题组[32]扩展为选定药物激活免疫原性细胞死亡(ICDs)的能力,从而刺激抗肿瘤免疫反应。据报导[33],特定药物(如吉西他滨)可以刺激巨噬细胞的细胞毒潜能和M1样分化。在TAMs对化疗的消极作用中,TAMs可以通过以下3个机制降低化疗药物的疗效:1)增加免疫抑制髓系细胞的募集;2)抑制适应性抗肿瘤免疫反应;3)激活癌细胞中的抗凋亡程序[34]。临床前研究[35]发现,在耐药肿瘤中经常观察到巨噬细胞募集的增加,这提示我们耐药的一个机制是TAMs的募集。在胶质母细胞瘤患者中,对贝伐单抗的耐药性是由于肿瘤边缘巨噬细胞迁移抑制因子(MIF)表达减少,导致M2巨噬细胞增多,因此促进了肿瘤生长。同样,肿瘤相关巨噬细胞分泌MMP9与抗血管内皮生长因子和抗胎盘生长因子药物阿柏西普的耐药性有关[36]。与源自骨髓和乳腺癌细胞系的巨噬细胞的体外共培养研究表明,巨噬细胞导致了紫杉醇、阿霉素和依托泊苷的化疗耐药[37]。在小鼠肿瘤模型和患者的乳腺癌组织中,用紫杉醇治疗的肿瘤比未用紫杉醇治疗的肿瘤显示更高的TAMs浸润,TAMs浸润是由暴露于紫杉醇后肿瘤细胞中升高的CSF-1 mRNA表达介导的,招募的TAMs抑制了紫杉醇诱导的有丝分裂阻滞,从而导致耐药的产生。通过阻断CSF-1-CSF-1受体(CSF-1R)信号传导来抑制TAMs募集,可以增强紫杉醇作用,并延长小鼠的生存期[38]。阿霉素治疗的乳腺癌患者表现出基质细胞产生CCL2的增加以及随后CCR2+单核细胞的募集,这与肿瘤复发有关[39]。在结直肠癌中,TAMs在5-氟尿嘧啶(5-FU)治疗期间分泌腐胺抵抗5-FU的疗效[40]。TAMs来源的外泌体(MDE)可引起胰腺导管腺癌(PDAC)对吉西他滨的耐药,缺失Rab27A/B基因以抑制外泌体分泌可以提高PDAC对吉西他滨的敏感性[41]。
接受X-射线放射治疗的胶质母细胞瘤显示巨噬细胞普遍减少,TAMs的M2/M1比值增加,这一影响可以通过M2比M1巨噬细胞有对辐射的更高抵抗力来解释[42]。常规放疗(RT)照射的巨噬细胞会导致癌细胞侵袭和血管生成[43],用氯膦酸盐脂质体预处理荷瘤小鼠以清除肿瘤相关巨噬细胞的RT结果有所改善[44]。而低剂量的RT可将巨噬细胞重编程为iNOS+/M1表型,并通过NO依赖的机制介导效应T细胞重新聚集到肿瘤组织而产生有效的肿瘤免疫排斥反应[45]。据报道,在乳腺肿瘤中,TAMs分泌的IL-4限制了放射治疗的疗效[46]。Xu等[47]研究发现,前列腺癌患者血清中的CSF-1水平在放疗后增加,CSF-1R的选择性抑制剂与放疗联用比单独放疗能更有效地抑制肿瘤的生长。
3 靶向治疗策略
TAMs在肿瘤发生、发展中发挥着重要作用,引发研究者们对肿瘤与TAMs关系研究的持续深入。目前针对TAMs的治疗方法主要包括:1)抑制TAMs的募集;2)清除TAMs;3)重新编程TAMs;4)调节TAMs吞噬作用(图2)[48]。

图2 靶向TAMs的免疫治疗策略[48]Fig.2 Immunotherapeutic strategies targeting TAMs[48]
3.1 抑制TAMs募集
肿瘤细胞可以通过分泌许多趋化因子招募巨噬细胞进入肿瘤组织。巨噬细胞被招募到肿瘤组织后,肿瘤细胞分泌各种细胞因子、代谢产物和外泌体改变TAMs的功能并使之极化。CSF-1和CCL2都负责将TAMs招募到TME中。据报导[49],在小鼠乳腺癌模型中,CCL2可阻断单核细胞从骨髓向血液的动员。通过抗CCL2单克隆抗体(如carlumab)或抑制其合成(如bindarit、 trabectedin)可抑制CCL2,阻止巨噬细胞聚集到肿瘤部位。Carlumab是一种抗CCL2的人单克隆抗体,曾安全地用于治疗转移去势性前列腺癌[50],并在一项Ib期研究中与其他化疗药物(紫杉醇、多西他赛、卡铂、吉西他滨和聚乙二醇化脂质体阿霉素)联合使用[51]。然而,也有研究表明,CCL2抑制的中断会导致过度转移并加速死亡,这是骨髓中单核细胞释放和原发肿瘤中癌细胞动员增强的结果,以及以白介素6和血管内皮生长因子A依赖的方式,在肺内形成血管和促进转移细胞增殖的结果[52]。除了针对CCL2的单抗外,其他化合物(如bindarit、 trabectedin)也被发现能抑制CCL2/MCP-1的合成。Bindarit可以减少TAMs和骨髓源性抑制细胞浸润,并可以抑制小鼠前列腺癌细胞转移[53]。BMS-813160是CCR2/CCR5双重拮抗剂,可抑制调节性T细胞和髓源性抑制细胞浸润,已经在非小细胞肺癌、肝细胞癌和胰腺导管腺癌(NCT04123379、NCT03496662)的联合治疗中试验了其治疗效果[54]。科学家们[55-57]还提出集中抑制CSF-1/CSF-1R信号轴的治疗策略,针对CSF-1(抗CSF1抗体)或CSF1-R(BLZ945、emactuzumab、PLX3397)的单克隆抗体和小分子进行了大量研究,并被证明以组织特异性的方式清除巨噬细胞。在这些小分子中,Pexidartinib(PLX3397)作为一种口服酪氨酸激酶抑制剂,2019年被美国FDA批准用于治疗腱鞘巨细胞瘤[57]。BLZ945是一种高度选择性的CSF1-R酪氨酸激酶小分子抑制剂,在小鼠宫颈癌和乳腺癌模型中降低TAMs的周转率,同时增加了浸润宫颈癌和乳腺癌的CD8+T细胞的数量[55]。此外,CSF-1R抑制剂SNDX-6352与派姆单抗联合正在进行第二阶段试验(NCT04301778)的评估。然而,不良反应限制了CSF-1R抑制剂的临床应用,Pexidartinib的副作用包括天冬氨酸氨基转移酶水平升高、疲劳、恶心和潜在的肝脏毒性等[58-60]。最近,一种MET/CSF-1/SRC抑制剂TPX-0022在具有MET基因改变的晚期实体瘤患者中进行了1期试验(NCT03993873)。另外,AMG820、RG7155、GW2580等几种小分子也在临床试验中[60-61],在某些情况下,高浓度CCL5也可通过与单核细胞表面的CCR2连接而导致TAMs的募集。吉非替尼作为一种酪氨酸激酶抑制剂,可以减少CCL5的分泌,抑制TAMs和前列腺癌细胞之间的串扰,导致肿瘤细胞的增殖和抑制多西紫杉醇活性[62]。此外,CXCL12/CXCR4信号级联促进癌症进展和转移并调节TAMs募集[63-64],因此,阻断CXCL12/CXCR4轴也可能是抑制TAMs招募的一种策略。一项研究表明,抑制CXCL12/CXCR4轴可以抑制TAMs的聚集和脓毒症后诱发的小鼠肿瘤进展[65]。在一些针对卵巢癌或前列腺癌的临床前研究中,阻断CXCL12/CXCR4信号通路可以延长小鼠的存活时间[66]。此外,最近一项研究报导了C-X3-C基序趋化因子配体1(CX3CL1)/CX3C基元趋化因子受体1 (CX3CR1)轴通过调节TAMs的募集来促进皮肤癌变[67],因此,抑制CX3CL1/CX3CR1轴也为抑制巨噬细胞募集提供了新的可能。
3.2 清除TAMs
除了抑制TAMs这一策略外,清除TAMs也是靶向策略之一。已被用作抗肿瘤药物的Trabectedin是一种四氢异喹诺酮类生物碱,它对癌细胞的作用机制很复杂,因为它与DNA共价结合,阻断转录,干扰DNA修复效率,使同源重组缺陷的癌细胞对这种治疗特别敏感。Trabectedin还可以使DNA双链断裂和细胞周期阻滞,从而导致癌细胞死亡[68]。在接受Trabectedin治疗的肿瘤患者中,观察到对包括TAMs在内的单核细胞的细胞毒性是其抗肿瘤活性的关键[69]。双膦酸盐是一类结构上由2个磷酸基团连接的中心碳组成的化合物,有R1和R2侧基团,R1是H、OH或Cl, R2由不同的官能团组成,决定化合物的效能[70]。双膦酸盐对羟基磷灰石有很高的亲和力,常用于骨质疏松症、变形性骨炎和骨转移等骨病的治疗[71]。在小鼠乳腺肿瘤模型中的临床前研究表明,双膦酸盐也显示出一定的抗肿瘤活性,且其抗肿瘤活性由TAMs介导[72]。为了改善药代动力学,减少毒副作用,双膦酸盐通常被配制成脂质体或纳米颗粒,从而改变生物分布,使其远离骨骼,用于骨外应用[73]。脂质体包裹的氯膦酸盐可以耗竭体内巨噬细胞,并在临床前模型中被证明能够介导巨噬细胞依赖的血管生成、肿瘤负担和转移的减少[74]。然而,使用氯膦酸脂质体不加选择地清除全身巨噬细胞,有时可能加剧疾病进展。例如,在B16F10肿瘤模型中,肿瘤引流淋巴结被膜下窦CD169+巨噬细胞的消耗与肿瘤负荷增加有关,而结直肠癌和乳腺癌患者淋巴结CD169+巨噬细胞的密度也与预后良好呈正相关[75-77]。这些研究结果进一步表明,与一般的巨噬细胞清除策略相比,以TAMs为靶点的治疗方法更可取,因为它可以保留其他潜在有益的组织驻留巨噬细胞。其他清除巨噬细胞的机制包括达沙替尼(Dasatinib),一种针对表达MMP9的巨噬细胞的酪氨酸激酶抑制剂,目前用于治疗慢性粒细胞白血病[78]。福氏志贺菌是一种诱导巨噬细胞凋亡的细菌,由Galmbacher和他的同事开发,用来感染携带乳腺癌的小鼠,发现其导致TAMs耗尽,并进一步导致荷瘤小鼠肿瘤完全消退[79],此外,毒素偶联单抗还能直接杀死巨噬细胞。一种与皂苷毒素结合的抗清道夫受体A(CD204)抗体可减少卵巢癌临床前小鼠模型中血管白细胞的数量并抑制肿瘤形成[80]。还有一种策略是激活T细胞以选择性地消除TAMs。TAMs表达大量的legumain,这是一种有助于ECM降解和血管生成的天冬酰胺内肽酶。已证明针对legumain的免疫可导致TAMs的CD8+依赖性消除[81]。集落刺激因子(CSF)-1在单核巨噬细胞系统的成熟、分化和存活中起着至关重要的作用,阻断CSF-1/CSF-1R轴也会导致大量TAMs凋亡[82]。
3.3 重新编程TAMs
巨噬细胞具有可塑性,这使得它们能够改变自身在肿瘤微环境中的表型。因此,将TAMs从促肿瘤表型重新编程为抗肿瘤表型是一种有吸引力的治疗策略。这些药物包括CD40激动剂、TLR激动剂、PI3Kγ抑制剂和IIa类HDAC抑制剂。激活TAMs用于抗癌治疗策略之一是靶向CD40,CD40在B细胞、巨噬细胞、树突状细胞等抗原提呈细胞和肿瘤细胞上表达[83]。在一项胰腺导管腺癌的临床前研究中,CD40的激活将免疫抑制巨噬细胞逆转为免疫保护巨噬细胞,从而重新建立肿瘤免疫监测[84]。目前,CD40有3个单克隆抗体具有良好的抗肿瘤作用,分别是CHIR-12.12、SGN-40和CP-870、893[85-87],三者中CP-870、893可选择性结合CD40非配体结合位点,增强细胞因子如IL-12、IL-23和IL-8的分泌,增强免疫细胞杀伤活性[13]。CP-870、893的临床试验表明,其可作为单一药物或与化疗药物联合使用,对多种肿瘤类型有效[88]。Toll样受体(TLR)可以识别病原体相关分子模式 (PAMP)和危险相关分子模式(DAMP),这对于巨噬细胞的先天免疫反应是必不可少的。据报导,TLR 激动剂,主要是 TLR4、TLR7/8和TLR9激动剂,通过刺激 TAMs极化为M1促炎表型而用于癌症治疗[89]。例如,TLR9激动剂IMO-2125在小鼠模型中诱导抗肿瘤巨噬细胞极化和肿瘤消退,已在许多转移性黑色素瘤的临床试验中得到评估[90]。此外,Huang等[91]报道,用于癌症治疗的临床核酸药物递送的阳离子聚合物可以通过TLR4信号激活引起TAMs复极化,从而促进小鼠肿瘤模型中的抗肿瘤免疫。然而,通过TLR刺激激活肿瘤相关巨噬细胞会伴随着PD-L1表达的升高,从而抑制T细胞。为了克服这种补偿作用,将TLR7激动剂1V270与PD-1阻断剂一起进行测试,观察到1V270诱导M1/M2 TAMs比率增加并提高PD-1阻断剂在头颈部鳞状细胞癌HNSCC中的功效[92]。TLR7/8激动剂3M-052和NKTR-262在临床应用中与PD-1阻断联合使用,以延长治疗效果和持续时间[93]。PI3Kγ信号可以抑制NF-κB的激活,随后触发一个转录程序,促进肿瘤生长过程中的免疫抑制,因此,抑制PI3Kγ可能通过刺激T细胞活性恢复免疫监测并抑制肿瘤发展。在小鼠肿瘤模型中,使用PI3Kγ选择性抑制剂IPI-549对PI3Kγ进行药理抑制会导致巨噬细胞重新编程,从而减少促肿瘤巨噬细胞,同时增加抗肿瘤巨噬细胞和T细胞反应[94]。抑制IIa类HDACs是再教育巨噬细胞发挥抗癌作用的新兴策略。HDACs能够从含有赖氨酸残基的组蛋白和非组蛋白中去除乙酰基,从而参与基因表达的表观遗传调控,Guerriero等[95]和Zhang等[48]指出,选择性IIa类HDAC抑制剂TMP195的治疗可诱导TME中促炎巨噬细胞和吞噬巨噬细胞的募集和分化,并使巨噬细胞重新极化为抗肿瘤表型,从而降低肿瘤负担和转移。
除了以上治疗策略,还有研究证明,铜绿假单胞菌甘露糖敏感血凝素在用于恶性胸腔积液(MPE)治疗时可将CD163+TAMs再培养为M1型巨噬细胞,这一发现说明重编程CD163+TAMs可作为MPE的一种治疗手段[96]。枸橼酸焦磷酸铁注射剂在抑制小鼠皮下腺癌生长的同时伴随着肿瘤组织中促炎性M1 TAMs的增加[97]。胸腺肽-α可以将TAMs再培养成能产生高水平促炎细胞因子的树突状细胞来参与抗肿瘤宿主反应,胸腺肽-α纳米载体可以提高癌症患者的免疫活性,临床试验表明胸腺肽-α可延长转移性黑色素瘤和晚期非小细胞肺癌患者的生存期[98]。β-葡聚糖是来自酵母的多糖之一,已被证明可以使TAMs分化为M1表型,是一种具有抗癌特性的有效免疫调节剂[99]。一些天然产物也可以通过改变巨噬细胞的极化来抑制肿瘤生长。例如,来自绿籽莲胚的甲基荷花碱通过调节卵巢癌中TAMs的极化对血管生成发挥抗肿瘤作用[100];来自浆果的脱氧五味子素可以抑制M2巨噬细胞的活性;洋葱素A不仅可以增加对卵巢癌细胞的细胞毒性,还可以抑制M2巨噬细胞的激活[101];葛根素能抑制非小细胞肺癌移植瘤模型的生长,增加M1巨噬细胞,降低M2标志物,增强抗肿瘤细胞因子的表达水平,降低促肿瘤细胞因子的表达水平[102]。另外,运用纳米药物将TAMs重编程为M1表型的报导也有很多,例如Zanganeh等[103]发现ferumoxytol可以将M2型TAMs转换为M1型TAMs,并抑制原发和转移性肿瘤在肝脏和肺部的生长。Rodell等[104]发现,环糊精纳米粒能够将小分子Toll样受体7/8激动剂靶向巨噬细胞,通过诱导M2到M1的极化来提高检查点抑制免疫治疗的效果。Cao等[105]发现人参提取物纳米颗粒可以通过激活TLR4将TAMs重新极化为M1巨噬细胞。Han等[106]制备的双靶向纳米复合物可以促进黄芩苷、抗原和免疫刺激传递到M2型 TAMs,从而极化和逆转M2 TAMs表型,重塑肿瘤微环境,并杀死肿瘤细胞。此外,活性microRNAs (miRNAs)作为转录和翻译的重要调控因子对TAMs极化也有显著影响。耗尽巨噬细胞中的miRNA加工酶Dicer会导致miRNA活性的破坏,从而促使M1型TAMs编程,增加CTLs在肿瘤中的浸润[107]。
3.4 调节TAMs吞噬作用
巨噬细胞可以利用其吞噬作用清除病原体和凋亡细胞,有时它的吞噬活性也会被某些信号抑制,如“不要吃我”信号。“不要吃我”信号与巨噬细胞上的大多数细胞表面蛋白相互作用,抑制巨噬细胞的吞噬作用。Majeti等[108]和Barkel等[109-110]确定了3种抑制吞噬作用的途径,分别是信号调节蛋白α(SIRPα)/CD47途径,主要组织相容性复合体I类/白细胞免疫球蛋白(Ig)样受体亚家族B成员1(MHC-1/LILRB1)途径,以及CD24/唾液酸结合Ig样凝集素10(Siglec-10)途径。SIRPα/CD47信号通路是该团队发现的3个通路中的第1个,巨噬细胞表达信号调节蛋白α(SIRPα)是一种识别CD47的受体,CD47是一种“不要吃我”信号,在不同类型的肿瘤细胞上普遍表达,并与SIRPα结合,提供下调信号,阻止肿瘤细胞吞噬[111]。因此,通过抗CD47抗体、工程化诱饵受体、抗SIRPα抗体或双特异性抗体药物阻断CD47/SIRPα轴,可恢复巨噬细胞对肿瘤细胞的吞噬功能并激活抗肿瘤免疫反应[112]。一些靶向该途径的药物已广泛用于抗肿瘤治疗,大多数已进入临床试验[113-115]。Ko等[116]设计一种溶酶体酶激活的vSIRPα探针,能够促进CD47靶向的癌症成像,并根据吞噬作用引发抗癌免疫应答。全身给药后,该探针显示出明显的肿瘤靶向性,为肿瘤治疗提供一种新的策略。相似地,肿瘤相关巨噬细胞表达的PD-1可与肿瘤细胞上的PD-L1相互作用,从而以SIRPα依赖的方式抑制吞噬[117]。Barkal等[109]发现的第2种抑制吞噬途径是MHC-1/LILRB1途径,巨噬细胞上表达的抑制性受体LILRB1(白细胞免疫球蛋白样受体B1)可以与肿瘤细胞上的MHC I类分子的β2-微球蛋白结合,从而抑制吞噬功能。研究人员评估了各种人类癌细胞系在抗CD47治疗后人类巨噬细胞的吞噬作用,发现一些细胞系在抗CD47治疗后没有表现出明显的吞噬作用,然后它们产生MHC-1抗体,一种阻断人类白细胞抗原A、B和C(HLA-A、HLA-B、HLA-C)的抗原结合片段(Fab),并使用这种抗体来阻断LILRB1。在选择SIRPα/CD47阻断表现出强耐药性的细胞系后,他们发现阻断MHC-1/LILRB1通路可以在体内体外促进吞噬作用。Barkal等[110]发现的第3条抑制吞噬途径是CD24/Siglec-10途径,CD24作为卵巢癌和乳腺癌的主要先天免疫检查点,是癌症免疫治疗的理想靶点。它通过结合一种由TAMs表达的抑制性受体Siglec-10来抑制吞噬作用,Barkal等[118]发现敲除CD24、Siglec-10基因和利用单克隆抗体的基因敲除来阻断CD24/Siglec-10轴,可以增加巨噬细胞吞噬肿瘤的能力,并减缓体内巨噬细胞依赖性肿瘤的生长。
4 TAMs靶向治疗与其他疗法联用
单一的TAMs靶向疗法已经在肿瘤治疗中显示出疗效,而将TAMs靶向疗法与其他疗法联用的治疗方法也在研究中,这些疗法包括化疗、放疗和免疫检查点抑制剂,TAMs靶向疗法可以改善患者的预后,因此可以作为肿瘤治疗的补充治疗[48]。免疫检查点疗法目的是逆转肿瘤微环境的免疫抑制特性,恢复细胞毒性免疫细胞的抗癌功能。经过临床验证的检查点靶点有PD1、PD-L1和CTLA4,它们的抑制在黑色素瘤和霍奇金淋巴瘤等多种癌症治疗中发挥了显著的抗肿瘤作用[119]。然而免疫检查点抑制仅对部分患者有效,许多病例表现出无效性,甚至产生耐药性[120]。
巨噬细胞表达PD-1和CTLA4的配体(PD-L1、PD-L2、B7H4、B7-1和B7-2),有助于巨噬细胞发挥免疫抑制功能。TAMs靶向治疗可以补充免疫检查点阻断治疗的抗肿瘤作用,当与CSF-1R拮抗剂、PI3Kγ抑制剂、IIa类HDAC抑制剂或抗CD47抗体联合使用时,检查点阻断治疗显示出增强的杀瘤效果或恢复传统疗法耐药患者治疗反应的能力[48]。在一种侵袭性的乳腺癌转基因小鼠模型中,化疗联合阻断TAMs的方法可促进抗肿瘤免疫和细胞毒性T细胞的浸润,与单纯化疗相比,肺转移显著减少,总存活率提高[49]。此外,针对前列腺癌的临床前研究发现,CSF-1/CSF-1R通路与放射治疗疗效的极限有关。放射治疗和CSF-1R抑制剂联用比单纯放疗能更有效抑制肿瘤生长[47]。
5 总结与展望
在肿瘤微环境中,肿瘤细胞与TAMs之间存在着复杂的相互作用,大多数患者肿瘤中TAMs的高密度与不良预后相关,使TAMs成为肿瘤治疗的靶点。抑制TAMs募集、清除TAMs、TAMs重编程、调节巨噬细胞的吞噬作用可有效抑制肿瘤进展并改善癌症患者的预后。TAMs靶向疗法与其他方法联用也对癌症治疗效果有所改善。然而,TAMs靶向疗法仍然存在一些待解决的问题。一是小鼠和人巨噬细胞的差异。细胞表面标记F4/80的表达只在小鼠巨噬细胞中发现,而在人类细胞中检测不到[121];小鼠和人巨噬细胞在体外暴露于刺激性细胞因子后的转录图谱也存在差异,例如,小鼠巨噬细胞对免疫抑制的M2型的极化通常是通过IL-4或IL-13刺激来实现,这导致M2相关标记物的上调,包括FIZZ1、ARG1和YM1,而人类M2巨噬细胞中没有观察到这种反应;同样,NOS2和ARG1对精氨酸的竞争代谢被用来区分小鼠巨噬细胞中促炎症的M1和免疫抑制的M2内型,但这个判别标准不适用于人类细胞。研究者应认识到这些差异,并解决由小鼠实验向人体实验转化的问题。另外,由于肿瘤微环境中的巨噬细胞并不只是单纯的M1、M2型,识别主要由M1或M2产生的TAMs特异性标记物或分子将有助于开发更复杂的治疗方法,这种疗法可以针对肿瘤而不影响其他组织驻留免疫细胞的功能[14]。二是靶向巨噬细胞的副作用。因巨噬细胞在维持体内平衡方面的多方面作用,巨噬细胞的全身性耗尽可能导致感染增加或组织驻留细胞执行其正常功能的能力受损[122]。TAMs重编程是一个更好的选择,但杀瘤和促瘤功能之间的微妙平衡也需要好好把握。针对巨噬细胞重新编程的策略也应该旨在保护这些细胞在非肿瘤组织中进行吞噬和伤口愈合的能力。
巨噬细胞靶向药物主要包含抗体和小分子抑制剂两类,这两类药物在药理学特性上差异极大,相对分子质量较大的单克隆抗体通常对组织穿透效率较低,可延长肿瘤在血液中的滞留和清除。然而,小分子抑制剂比单克隆抗体特异性低,毒性风险却更高[123]。纳米颗料在载药方面的应用可以改善这个问题,研究人员也利用:1)巨噬细胞作为药物载体;2)巨噬细胞衍生的外泌体作为药物载体;3)巨噬细胞膜包裹的纳米粒作为药物载体。虽然将巨噬细胞工程用于癌症免疫治疗和药物输送方面取得一些成功,但挑战依然存在:首先,肿瘤微环境中巨噬细胞募集和极化的机制尚不完全清楚;其次,肿瘤组织的免疫微环境很复杂,单靠巨噬细胞介导的癌症免疫疗法不足以根除肿瘤;再者,目前巨噬细胞载体靶向实体肿瘤组织的能力仍然有限[124]。最近开发一种新的基于巨噬细胞的细胞治疗技术:用CAR分子装备巨噬细胞。嵌合抗原受体(CAR)T细胞疗法已成功用于血液恶性肿瘤,然而其在实体瘤中的应用却不尽人意[125]。CAR NK 细胞治疗优于CAR T细胞疗法,但仍存在与CAR T治疗类似的局限性。TAMs在肿瘤微环境中大量存在,而且即使是免疫抑制的M2巨噬细胞也有很强的吞噬活性,这使得CAR M成为过继免疫疗法的一种替代疗法[126]。
据报导,Klichinsky等[127]用靶向HER2的CAR转染的巨噬细胞可以减轻肿瘤负担并延长生存期。然而CAR·M疗法还处于研究阶段,仍有许多挑战。我们可以相信,随着研究者们对肿瘤微环境中肿瘤相关巨噬细胞认识的继续深入,可以开发出更多有效的癌症治疗方法,但这还需要研究者们的持续努力。
