运动干预NAFLD的分子机制研究述评
——基于ROS调节UPRmt的线粒体毒性兴奋效应
2022-10-15丁树哲
张 媛,文 立,丁树哲
(1.南京体育学院 运动健康学院,江苏 南京 210014;2.上海交通大学 系统生物医学研究院运动转化医学中心,上海 200240;3.华东师范大学 青少年健康评价与运动干预教育部重点实验室,上海 200241;4.华东师范大学 体育与健康学院,上海 200241)
非酒精性脂肪性肝病(nonalcoholic fatty liver disease,NAFLD)是被严重低估的重大健康威胁,除会引发肝脏严重病变乃至癌变之外,还可能引发心血管及代谢疾病(Es‐tes et al.,2018)。据报道,由于生活方式的明显改变,全球NAFLD患病率在近几十年迅速增长。我国近1/3的人群患有NAFLD,其中10%~20%将发展为非酒精性脂肪性肝炎(non alcoholic steato hepatitis,NASH),而 NAFLD发展为NASH为不可逆过程(Tarantino et al.,2020)。预计到2030年,我国NAFLD患者将达到3.15亿,脂肪肝将成为我国慢性疾病防控中的最大负担(Byrne et al.,2015;Tanaka et al.,2019)。尽管NAFLD的病理现象被高度关注,但目前尚无美国食品药品监督管理局(Food and Drug Administration,FDA)批准的治疗NAFLD的药物。运动是防治NAFLD经济有效的干预方式之一,有研究证实,在NAFLD诱发进程中进行有效的运动、膳食干预对逆转NAFLD至关重要。有氧运动、抗阻运动或是简单的身体锻炼均可改善NAFLD(Hajighasem et al.,2019;Hashida et al.,2017;Oh et al.,2017)。运动不仅可以改善肝功能,降低NAFLD由轻度向重度转化的系统标志物水平,是治疗和预防NAFLD经济有效的干预方式,而且可以降低患心血管疾病、糖尿病及高血压的风险(Farzanegi et al.,2019;van der Windt et al.,2018)。氧化应激与NAFLD密切相关,线粒体活性氧(reactive oxygen species,ROS)生成增多可改变细胞氧化应激水平,过高ROS会扰乱线粒体蛋白环境,加剧线粒体损伤。而适量ROS对激活细胞和有机体的适应性反应有益,这种剂量效应表现为线粒体毒性兴奋效应。本文围绕NAFLD发展过程中氧化应激水平改变及其调控的线粒体未折叠蛋白反应(mitochondrial unfolded protein response,UPRmt),综述运动预防、逆转NAFLD的生物学机制,揭示线粒体毒性兴奋效应在预防NAFLD发生发展中的规律,为NAFLD的防治提供理论依据。
1 氧化应激与炎症是NAFLD发展的核心环节
目前,NAFLD的发病机制尚不完全清楚。研究表明,其与多种继发性并发症密切相关,如胰岛素抵抗(insulin resistance,IR)、肥胖、高血压、血脂异常、2型糖尿病(type 2 diabetes mellitus,T2DM)及心血管疾病等,其中肥胖是最重要的NAFLD诱发因素(Neuschwander‐Tetri,2017;Sar‐war et al.,2018)。传统的“两次打击”理论,即第一次打击为肝脏脂质沉积和胰岛素抵抗,第二次打击为氧化应激,其在一定程度上解释了NAFLD的发病机制(Rolo et al.,2012;Serviddio et al.,2013)。之后,“多重打击”理论即从遗传易感性、表观遗传、肝细胞内代谢、肝细胞内的细胞互作、脂肪组织等多方面考虑了诸多因素并行的作用效果,其中,氧化应激被认为是造成“多重打击”的重要诱因,是导致NAFLD发展中肝脏损伤的主要因素(Fried‐man,2018)。氧化应激反应是细胞内ROS与抗氧化系统清除能力不平衡的结果(Takaki et al.,2013)。肝内脂质超载可激活多个ROS生成通路导致氧化物过量生成,高水平ROS影响细胞大分子(DNA、脂类、蛋白质等)的氧化修饰,致使大分子损伤积累,引起肝损伤(Rolo et al.,2012;Serviddio et al.,2013)。另一方面,ROS信号可能在正常生理过程中发挥重要作用,如调节细胞内稳态或参与稳态应激反应、代偿性适应代谢功能障碍和炎症反应等(Forrester et al.,2018;van der Vliet et al.,2018)。可见,ROS信号介导的NAFLD发生发展机制及特异性分子途径仍不清晰,有待进一步研究。
1.1 肝脏脂质代谢异常与氧化应激
脂肪代谢不平衡是诱发NAFLD的直接病因,阐明肝脏能量代谢底物——游离脂肪酸(free fatty acids,FFAs)的来源及去路是深入了解NAFLD潜在致病机制的根源(Chen et al.,2019)。研究表明,导致肝脏脂肪沉积的FFAs约超过一半来自外周脂肪分解或未酯化脂肪酸(nonester‐ifed fatty acid,NEFA)库(Arab et al.,2018)。胰岛素可调节脂肪组织甘油三酯分解,NAFLD中外周组织胰岛素敏感性和葡萄糖利用率较低,而IR并未降低脂肪组织的脂解作用,因此导致NEFA过度分配至肝脏(Samuel et al.,2018)。肝脏FFAs第二大来源为脂肪从头合成(de novo lipogenesis,DNL),即由肝脏细胞将多余葡萄糖和果糖转化为脂肪酸的过程。Donnelly等(2005)的同位素标记实验发现,NAFLD中肝脏增加的脂肪主要来自于DNL作用。Samuel等(2018)研究发现,骨骼肌IR会导致高血糖症和高胰岛素血症,两者作用可激活肝细胞碳水化合物反应元件结合蛋白(carbohydrate response element binding protein,ChREBP)及转录因子固醇调节元件结合蛋白1(sterol regulatory element binding protein 1,SREBP‐1)活性,提高脂肪合成酶表达水平,使肝脏合成FFAs增加。另一方面,FFAs的清除是决定FFAs稳态的另一重要环节。肝脏FFAs主要通过线粒体β氧化和再酯化合成甘油三酯进一步代谢(Arab et al.,2018)。甘油三酯是形成肝脏脂滴的主要脂类物质,能够以极低密度脂蛋白(very low densi‐ty lipoprotein,VLDL)的形式输出(图1)。
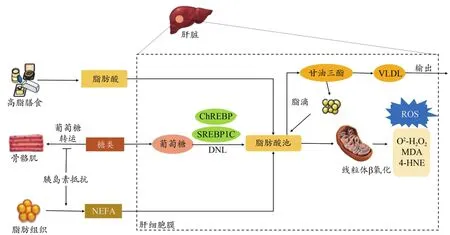
图1 NAFLD脂代谢与氧化应激Figure 1.Lipid Metabolism and Oxidative Stress in NAFLD
过量FFAs将作为底物生成脂毒性物质,如神经酰胺、甘油二酯及溶血卵磷脂等,最终引起代谢应激、炎症反应及细胞死亡(Friedman et al.,2018;Chen et al.,2019)。氧化应激是体内氧化与抗氧化作用失衡的一种状态。ROS是不同类型肝细胞内代谢产生的副产物,包括不断生成的超氧化物阴离子(O·)和过氧化氢(HO)等物质(Ma‐sarone et al.,2018)。ROS含量过高可使肝内抗氧化系统遭到破坏。临床与动物实验通常将血液或肝脏组织中氧化应激与抗氧化物的标志性产物水平作为评价NAFLD/NASH氧化应激状态的依据。氧化应激标志物主要包括脂质损害产物硫代巴比妥酸反应物(thiobarbituric acid reac‐tive substance,TBARS)、丙二醛(malondialdehyde,MDA)、4-羟基壬烯醛(4‐hydroxynonenal,4‐HNE)、氢过氧化物和 8-异前列烷、DNA氧化产物(8-羟基脱氧鸟苷)、蛋白质氧化产物(蛋白质羰基、硝基酪氨酸)等,抗氧化物主要含有谷胱甘肽(glutathione,GSH)、抗氧化酶如超氧化物歧化酶(superoxide dismutase,SOD)、过氧化氢酶(catalase,CAT)以及谷胱甘肽过氧化物酶(glutathione peroxidase,GSH‐Px)等。
1.2 NAFLD发展过程中线粒体功能变化
肝细胞的能量稳态主要受线粒体氧化代谢调节,包括β氧化、三羧酸循环、生酮作用、电子传递链(electron transport chain,ETC)活性与 ATP 合成等(Sunny et al.,2017)。NAFLD早期肝细胞内FFAs内流迅速增加,引起多种激素和代谢发生改变,增强线粒体脂肪酸氧化(fatty acid oxidation,FAO)作用,是代偿性降低肝脏脂肪积累的适应性表现。大多研究表明,营养过剩的小鼠模型中肝脏线粒体β氧化显著增高,而有些研究结果出现差异性的原因可能在于高脂膳食的饮食成分、干预时长、小鼠基因背景以及检测FAO的方法有所不同。另外,NAFLD病情发展程度不同,线粒体功能亦有所差异。Kakimoto等(2019)研究表明,肝脏线粒体FAO能力在高脂膳食(high fat diet,HFD)喂养4周时有所降低,而在喂养8周时却得以恢复,提示高脂膳食诱导代谢受损的肝脏线粒体功能具有重塑性。而NAFLD初期肝脏脂肪变性所伴随的高水平线粒体FAO机制还有待进一步研究。目前研究提示,高水平线粒体FAO可能与肉毒碱棕榈酰转移酶1(carnitine palmitoyltrans‐ferase‐I,CPT‐1)活性及过氧化物酶体增殖激活受体 α(per‐oxisome proliferator‐activated receptor α,PPARα)表达水平增高、肝脏IR的发生、瘦素等激素水平增高以及FGF21有关(Satapati et al.,2012;Schuster et al.,2018)。
研究表明,不同NAFLD或NASH阶段的肝脏线粒体ETC复合物活性、耗氧率、氧化磷酸化(oxidative phosphor‐ylation,OXPHOS)效率以及线粒体膜电位呈现不同变化特征。导致这些差异的原因可能与疾病病程肝脏氧化应激程度不同有关,如Koliaki等(2015)发现,肝脏线粒体OXPHOS效率在简单脂肪变性机体中显著增高,而在NASH患者中显著下降。基因瘦素缺陷ob/ob小鼠和瘦素抵抗db/db小鼠以中度肝炎症和轻度纤维化为特征(Fried‐man et al.,2018),其肝脏线粒体氧化能力有所增强,而ETC复合物活性被显著抑制,NASH小鼠的肝脏线粒体ETC明显受损(Cheng et al.,2009;Finocchietto et al.,2011)。由此得出:ETC活性伴随NAFLD发展逐渐下降,而线粒体FAO作用在NAFLD和NASH中均显著增高。这种在NAFLD发展过程中出现的线粒体FAO作用代偿性增强,而ETC并未随之上调,即FAO与ETC不协调的现象最终导致更多底物衍生的还原当量进入受损ETC产生电子漏,致使ROS生成增多(Begriche et al.,2013)。
1.3 氧化应激与线粒体功能的恶性循环
细胞、动物、人体等不同水平研究表明,线粒体ROS水平增高是线粒体功能受损,导致NAFLD的主要原因之一(Farzanegi et al.,2019;Simoes et al.,2018;Spahis et al.,2017)。线粒体ROS释放可通过磷酸化和转录因子等多种修饰作用调节胞质内氧化应激环境,以维持线粒体完整性和生物稳态(Forrester et al.,2018;Vercesi et al.,2018)。不同ROS水平可调节不同类型信号分子,从细胞生物适应到细胞死亡(Figueira et al.,2013),高水平ROS会导致大分子非特异性受损,通常将产生更多中间产物并触发一系列放大损伤效应,加快疾病进展(Rolo et al.,2012;Serviddio et al.,2013)。
脂质过氧化是NASH发展的主要诱因之一。研究表明,NAFLD患者的循环血液中已出现多个脂质过氧化的生物标记物,如MDA、4‐HNE,其浓度与NAFLD发展程度成正相关(Liu et al.,2011;Svegliati‐Baroni et al.,2019),与 NASH 组织学特征高度相关(Albano et al.,2005;Bel‐lanti et al.,2017)。有研究发现,脂质过氧化物可损伤线粒体膜,使正常ETC受损(Masarone et al.,2018)。此外,作为线粒体内膜的重要磷酸分子——心磷脂,对氧化损伤十分敏感,极易改变线粒体膜的流动性与稳定性,导致ETC复合物活性下降(Li et al.,2010),为触发caspase介导的细胞凋亡通路提供条件(Kagan et al.,2005)。除了脂类物质的影响,线粒体内膜生成的O·和HO也较易与临近的线粒体DNA(mitochondrial DNA,mtDNA)相结合,导致DNA缺失和点突变,并加剧线粒体损伤(Borrelli et al.,2018)。因此,ROS引起的mtDNA受损与突变可改变ETC活性,触发线粒体ROS生成增多,使损伤程度进一步扩大。损伤后的线粒体ETC被抑制,同时膜结构完整性被进一步破坏,引发线粒体源性氧化应激的恶性循环,而终止这一恶性循环的关键环节是提供外源性干预,帮助线粒体恢复氧化应激稳态,重塑线粒体功能。
2 ROS调节UPRmt的线粒体毒性兴奋效应
2.1 诱发UPRmt的关键调控机制
线粒体依赖于一个复杂的蛋白质网,使其在细胞能量供应、代谢、凋亡等诸多方面发挥重要作用(Moehle et al.,2019)。线粒体蛋白质受细胞核与线粒体两套基因组编码,约1 100个线粒体蛋白中除13个受mtDNA编码外,绝大多数线粒体蛋白由细胞核编码,需通过线粒体蛋白输入机制进入线粒体内部(Zhang et al.,2020)。线粒体ETC在能量生成过程中产生的ROS,不仅可干扰OXPHOS过程,同时也会损害线粒体蛋白、脂质及mtDNA(Moehle et al.,2019)。为了建立与维持最佳蛋白环境,线粒体需要通过多钟机制维持蛋白环境稳态,若蛋白稳定防御途径受损将对机体健康产生重大影响。
UPRmt是由于线粒体蛋白稳态被破坏,发生多种蛋白毒性应激,最终启动相关基因表达程序,上调线粒体特异性伴侣蛋白和蛋白酶等靶基因,以恢复线粒体内蛋白稳态的过程(Yi et al.,2018)。UPRmt机制最初在哺乳动物中被发现,并一直被高度关注,之后在线虫中被充分研究。研究发现,激活转录因子4(activating transcription fac‐tor 4,ATF4)和激活转录因子 5(activating transcription fac‐tor 5,ATF5)是哺乳动物线粒体应激反应与UPRmt的关键调控因子(Fiorese et al.,2016;Quiros et al.,2017)。UP‐Rmt机制通过感知蛋白毒性应激开启线粒体-细胞核对话,诱导细胞做出适应性应答,从而改善蛋白折叠应激,重建线粒体蛋白稳态。哺乳动物的UPRmt与综合应激反应(integrated stress response,ISR)密切相关。线粒体蛋白稳态失衡使真核翻译起始因子2α(eukaryotic initiation fac‐tor 2,eIF2α)被磷酸化,进而导致蛋白质合成减少,但可通过调节ATF4、ATF5 mRNA中5’UTR非翻译区上游开放阅读框架(upstream open reading frames,uORFs),选择性启动ATF4、ATF5等基因翻译,随后转录调节多个线粒体伴侣蛋白和线粒体蛋白酶,提高线粒体蛋白含量折叠能力,恢复线粒体蛋白稳态(Melber et al.,2018)(图2)。
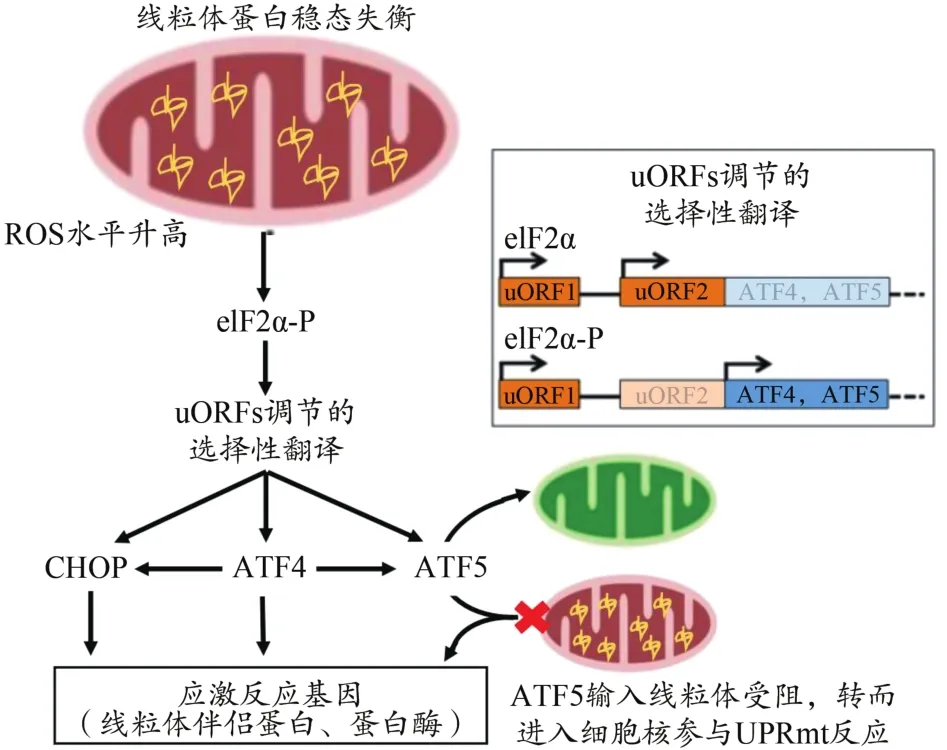
图2 哺乳动物UPRmt机制Figure 2.UPRmt Mechanism in Mammalian
2.2 UPRmt与线粒体毒性兴奋效应
生物体内的毒性兴奋效应(hormesis)指低剂量压力刺激可激活细胞和有机体的适应性反应以维持体内平衡,促进健康甚至延长寿命,而超过阈值的高剂量刺激则导致细胞损伤的一种现象,其与刺激源之间的关系不同于简单的线性关系,而通常表现为钟形或倒U性曲线。近年来,毒性兴奋效应的理论已扩展至线粒体水平。2006年,“线粒体毒性兴奋效应”(mitohormesis)的概念首次被提出(Tapia,2006;Yi et al.,2018),即对线粒体稳态的轻度扰动会促进与协调线粒体-细胞核之间的“对话”,降低细胞对外来刺激源的敏感性,提高细胞抵御刺激的能力。
多种刺激源可诱发线粒体产生毒性兴奋效应,例如线粒体ROS水平升高,ATP、Ca和NAD等代谢产物增加、线粒体蛋白稳态失衡等。其中,ROS生成是线粒体与细胞核“对话”的重要信号因子之一,也是调节线粒体毒性兴奋效应的主要信号。研究发现,ROS与UPRmt可能在诱发线粒体毒性兴奋效应中发挥协同作用,ROS‐UPRmt线粒体毒性兴奋效应信号对维持线粒体稳态至关重要(Monaghan et al.,2015)。Dillin等(2002)早期针对低等动物研究表明,扰乱核编码的线粒体OXPHOS复合物I、Ⅲ和Ⅳ,或通过限制葡萄糖摄入诱导低等动物体内ROS水平小幅度增高,均可激活UPRmt过程及线粒体毒性兴奋效应,延长蠕虫寿命。Gariani等(2016)同样发现,补充NAD生物合成的前体物质烟酰胺腺嘌呤二核苷酸可通过Sirt介导的UPRmt启动线粒体毒性兴奋效应,改善高脂高糖膳食小鼠的肝脏脂质沉积。此外,人体实验中,Ristow等(2009)发现,运动训练引起ROS水平适度增高,使机体胰岛素敏感性提高,而服用抗氧化剂对包括糖尿病在内的多种病理现象并无显著疗效(Kawagishi et al.,2014;Ristow et al.,2009)。因此,中度或短暂的线粒体应激将诱导蠕虫和哺乳动物发生UPRmt及线粒体毒性兴奋效应。这一机制可能以细胞自主和非自主的方式对宿主提供保护或发挥积极作用,对维持机体稳态至关重要。由此可见,研究UPRmt的调节机制对改善或预防代谢恶化及脂肪肝疾病的发生发展意义重大,深入探究UPRmt及线粒体毒性兴奋效应的上游调控机制可为探索人类脂肪肝疾病新治疗策略提供线索。
2.3 UPRmt激活与线粒体因子分泌
UPRmt的触发可能与组织或器官释放某些关键信号因子相关,即线粒体不仅通过UPRmt恢复稳态,同时也会发出“求救”信号,由发生线粒体应激的细胞释放一类核编码的信号分子至远处组织发挥代谢调节作用(Mottis et al.,2019;Yi,2019),这类分子被称为线粒体因子(mito‐kines)。线粒体与远处组织的通讯正是通过线粒体因子得以实现,但具体分子机制尚不清楚。
线粒体因子分泌水平与慢性肝病和心血管疾病风险密切相关,是预测NAFLD、NASH和晚期肝纤维化等肝脏疾病的标志分子(Conte et al.,2019;Yi,2019)。成纤维细胞生长因子 21(fibroblast growth factor,FGF21)与生长分化因子 15(growth differentiation factor15,GDF15)被认为是调节糖脂代谢以及胰岛素敏感性的内分泌因子,其分泌水平与线粒体功能状态密切相关(Conte et al.,2019;Klaus et al.,2020)。研究发现,FGF21和GDF15的分泌过程均依赖于激活elF2α磷酸化及其下游ATF4,而这一通路是激活 UPRmt的主要途径(Coll et al.,2019;Lewis et al.,2019;Munch,2018;Naresh et al.,2019)。在哺乳动物细胞中,线粒体的多种损伤(包括蛋白毒性应激和ROS)通过线粒体应激反应(mitochondrial stress response,MSR)发出信号。活化的一般性调控阻遏蛋白激酶2(general control nonderepressible 2 kinase,GCN2)可磷酸化elF2α,使蛋白质翻译过程减速,启动ATF4、ATF5和转录因子C/EBP的同源蛋白(C/EBP‐homologous protein,CHOP)表达(Munch,2018;Naresh et al.,2019)。在小鼠模型中,血液循环的FGF21和GDF15主要来源于肝脏组织的分泌,随后至外周组织,在受体细胞中激发特定效应。例如,Coll等(2019)在两项独立的随机对照临床试验中发现,二甲双胍可提高受试者循环GDF15水平,通过脑干组织中GDNF家族 α 样受体(GDNF‐family receptor α‐like,GFRAL)减少食物摄入并降低体质量,证实了血液循环中GDF15的水平对于机体维持能量平衡和体质量的重要性。
可见,UPRmt激活是线粒体因子分泌的诱因,是细胞应对线粒体应激引起能源危机的合理应答,对维持细胞代谢稳态至关重要。然而,UPRmt触发与线粒体因子分泌的关系仍不清楚,其分泌水平在UPRmt被激活的早期或晚期阶段所呈现的变化规律仍较模糊。
3 运动干预NAFLD的内在机制探讨
UPRmt与多种由蛋白毒性应激导致线粒体功能障碍的病理现象有关,包括NAFLD(Ortiz et al.,2020;Teodoro et al.,2013)。虽然UPRmt是NAFLD进展中维持与调节蛋白稳态的重要机制,但目前对临床前期与临床阶段患者的研究仍不够深入(Yi,2019)。Gariani等(2016)报道,高脂(44.6%)高糖(40.6%)膳食(hgh fat high sugar,HFHS)模型具有脂肪变性所致线粒体功能障碍的特点,并伴随炎症、纤维化标志物mRNA表达水平增高,但并未改变UPRmt标志物的mRNA水平,如酪蛋白水解蛋白酶P(ca‐seinolytic protease P,ClpP)、热休克蛋白 10(heat shock pro‐tein 10,Hsp10)和热休克蛋白 60(heat shock protein 60,Hsp60)等。有趣的是,补充NAD前体物质烟酰胺腺嘌呤二核苷酸可诱发UPRmt,并显著改善HFHS膳食引起的慢性肝脏脂肪异常沉积。这一研究提示,改变UPRmt水平是干预 NAFLD 的重要手段之一(Urbina‐Varela et al.,2020)。此外,研究发现,受损的UPRmt可导致肝细胞发生衰老,使肝硬化伴随代偿失常,在细胞培养实验中若通过恢复线粒体蛋白酶ClpP水平,上调UPRmt作用,即可激活线粒体毒性兴奋效应,从而防止肝细胞衰老,并有效改善肝细胞功能(Luo et al.,2021;Sen et al.,2019)。
肝脏氧化应激调节UPRmt的线粒体毒性兴奋作用,与肝脏脂质沉积密切相关。运动提高肝脏脂肪氧化水平,缓解脂质沉积是运动调节UPRmt的本质因素。运动对UPRmt的调节作用主要体现在运动改善肝脏氧化应激,增强线粒体-细胞核“对话”,促进线粒体因子分泌等方面(图3)。
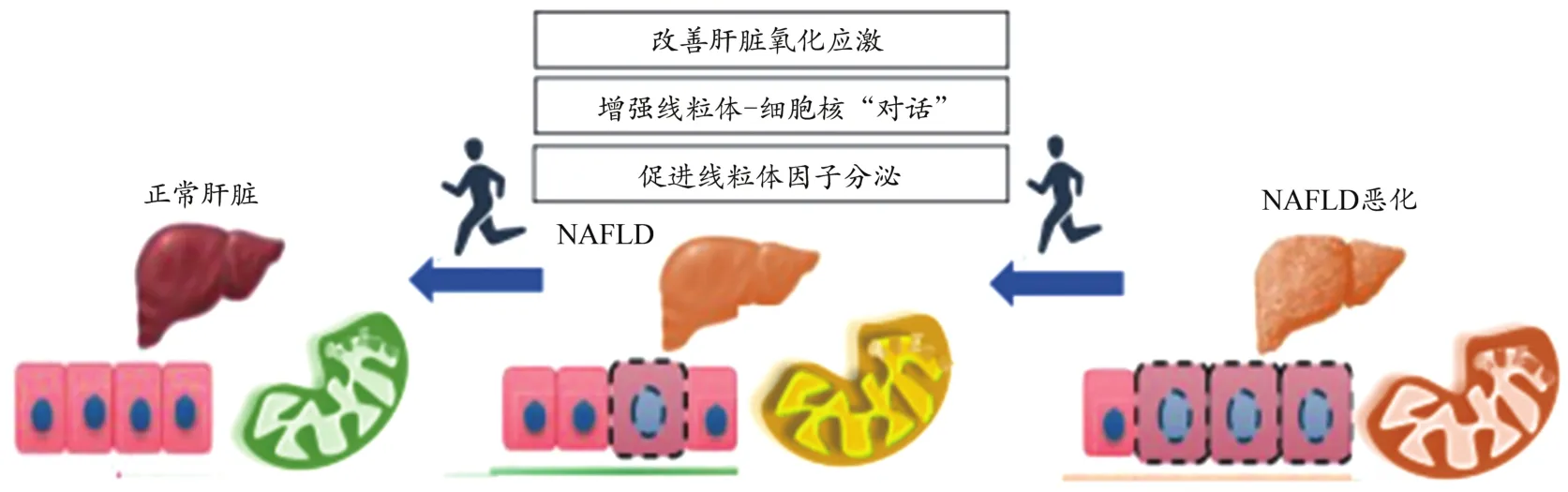
图3 运动调节UPRmt逆转NAFLD的内在机制Figure 3.Mechanism of Exercise Regulates UPRmt and Reverses NAFLD
3.1 运动降低肝脏脂质沉积,改善肝脏氧化应激水平
肝脏组织中氧化应激是导致肝细胞死亡和组织损伤的重要机制。NAFLD中肝脏线粒体异常,抗氧化酶下调,白细胞积累和肝脏炎症均是导致ROS生产过剩的主要原因。ROS过量产生不仅会引起脂质过氧化,导致炎症和纤维形成,而且抑制肝细胞分泌VLDL,进一步诱导肝脏脂肪堆积。运动改善肝脏氧化应激水平可通过缓解肝脏脂质沉积及增强肝脏细胞抗氧化能力两方面发挥作用。运动对肝脏脂质含量的调节作用体现在以下方面:1)运动提高机体胰岛素敏感性,减少FFA向肝脏的输入量,降低脂肪合成底物水平(Cuthbertson et al.,2016)。诸多人体实验表明,运动是改善IR缓解NAFLD的有效方法(Bacchi et al.,2013;Oh et al.,2015a,2015b;Sullivan et al.,2012;Sun et al.,2012)。2),运动下调SREBP‐1表达水平,降低脂肪合成酶活性,抑制肝脏脂肪合成(Cintra et al.,2018;Wu et al.,2015)。Oh等(2017)对中年肥胖男子进行不同强度、频率的运动干预研究发现,12周抗阻训练或高强度有氧训练干预均可使循环外周血液单核细胞SREBP‐1c表达降低,下调脂肪肝患者肝脏FFA从头合成。运动还可显著降低肝脏脂肪酸合成酶(fatty acid synthase,FAS)和硬脂酰辅酶A去饱和酶1(stearoyl‐CoA desaturase 1,SCD1)表达水平,使FFA水平下降,逆转肝脏脂肪变性(Suk et al.,2015;Tsuzuki et al.,2015;Wu et al.,2015),提高乙酰辅酶A羧化酶(acetylCoA carboxylase,ACC)磷酸化,抑制脂肪合成(Cho et al.,2014)。3),运动提高参与线粒体脂肪氧化的肉毒碱棕榈酰基转移酶-1(carnitine palmitoyltransferase‐1,CPT‐1)、乙酰辅酶 A 脱氢酶(acetyl CoA dehydrogenase,ACD)活性,加速脂肪分解,同时降低线粒体氧化应激水平,提高线粒体β氧化能力(Stevanovic et al.,2020;Zheng et al.,2019)。
运动不仅能够调节肝脏脂肪代谢水平,而且对机体抗氧化系统的调节作用比补充抗氧化剂的效果更佳。运动激活细胞ROS生成也是运动激活抗氧化系统的机制之一。运动可适度提高细胞ROS水平,通过多种途径激活抗氧化系统。运动还可激活腺苷酸活化蛋白激酶[ade‐nosine 5’‐monophosphate(AMP)‐activated protein kinase,AMPK]及沉默信息调节因子1(silent information regulator 1,SIRT1)通路(Cardaci et al.,2012),作用于过氧化物酶体增殖物激活受体-γ共激活因子-1α(peroxisome prolifer‐ators‐activated receptor γ coactivator 1α,PGC‐1α),上调抗氧化防御系统主要因子表达水平,如核因子E2相关因子2(nuclear respiratory factor 2,Nrf2),提高其转录调控的诸多抗氧化因子表达水平,包括:醌氧化还原酶1[NAD(P)H quinone oxidoreductase 1,NQO1]、血红素氧合酶1(heme oxygenase‐1,HO‐1)、SOD、CAT 及 GSH‐Px 氧化还原反应系统(Kasai et al.,2020;Vargas‐Mendoza et al.,2019)。Chartoumpekis等(2013)和Chowdhry等(2010)研究发现,在NAFLD模型中,敲除Nrf2基因的小鼠NASH发生率更高。相反,激活Nrf2可逆转高脂肪和高果糖食物喂养小鼠的IR、肝脏脂肪变性和纤维化(Sharma et al.,2018)。近期Dludla等(2020)研究发现,ROS抑制剂——N-乙酰半胱氨酸(N‐acetyl cysteine,NAC)可显著抑制NAFLD动物模型肝脏脂肪堆积,这一作用主要通过对脂肪酸膜转运蛋白CD36和转录因子SREBP‐1c/‐2及PPARα的调节作用。因此,通过运动干预调节抗氧化系统活性,改变氧化应激环境是改善NAFLD的有效途径之一。
3.2 运动维持线粒体蛋白稳态,增强线粒体-细胞核“对话”
作为细胞的能量和代谢中心,线粒体蛋白质稳态的失衡与线粒体功能障碍是衰老和衰老相关疾病发生的重要因素,线粒体蛋白稳态失衡与功能障碍的发生可能互为因果,但它们之间相互联系的具体机制尚不清楚(Andere‐asson et al.,2019;Li et al.,2019;Urbina‐Varela et al.,2020)。线粒体蛋白稳态维持受到线粒体蛋白输入机制(protein import machinery,PIM)、线粒体自噬及UPRmt等多方面影响(Zhang et al.,2020)。其中,UPRmt机制是通过感知蛋白毒性应激开启线粒体-细胞核“对话”,诱导细胞做出适应性应答,从而改善蛋白折叠应激,重建线粒体蛋白稳态的过程。目前,尚不清楚错误折叠或受损蛋白质如何被UPRmt识别。较为普遍的观点认为,激活转录因子 1(activating transcription factor 1,ATF1)是诱发 UP‐Rmt的“信使”,其对线粒体蛋白输入效率极为敏感,正常情况下,线粒体蛋白输入效率较高时,ATF1蛋白中N末端的靶向序列将新生的ATF1蛋白顺利输入至线粒体内部,随后被线粒体蛋白酶LONP降解。当线粒体处于应激状态时,蛋白输入效率的降低导致其在细胞质中大量堆积,ATF1蛋白C末端序列将ATF1输入至细胞核,进而激活使线粒体蛋白恢复稳态的相关转录基因表达,最终改善线粒体和细胞状态(Naresh et al.,2019)。
运动调节线粒体蛋白稳态的作用在骨骼肌组织中已被充分证实。早期研究发现,运动可调节PGC‐1α表达,增强线粒体-细胞核之间相互作用,协调线粒体生物发生(Safdar et al.,2011)。近些年,研究发现,衰老骨骼肌中线粒体蛋白稳态失衡是发生肌少症的主要原因之一,而运动可改善衰老骨骼肌线粒体蛋白稳态,延缓肌少症症状(Coen et al.,2019;Liu et al.2021;Picca et al.,2019)。由此可见,运动对维持线粒体蛋白稳态发挥积极作用。1)运动增加线粒体蛋白输入组件蛋白表达水平,使核编码的线粒体蛋白靶向性输入至线粒体不同区域发挥功能,这一发现在老年鼠实验中也同样被证实(Zhang et al.,2013)。2)运动提高线粒体自噬作用,促进线粒体更新,提高线粒体蛋白质量,缓解线粒体应激(Sorriento et al.,2021)。3)运动可调节线粒体-细胞核“对话”,上调UP‐Rmt标志性蛋白,激活UPRmt。运动对UPRmt的调节作用在许多组织中均有报道:Cordeiro等(2020)研究发现,有氧运动训练及高强度间歇训练可提高衰老小鼠的全身代谢水平,同时使线粒体与细胞核之间失衡,显著提高骨骼肌组织HSP60、LONP和线粒体蛋白酶Yme1L1等UP‐Rmt标志物含量。Braga等(2021)发现,运动可改变线粒体蛋白稳态,上调下丘脑UPRmt标志物表达水平及线粒体最大呼吸能力。Santos‐Alves等(2015)报道,运动可调节肝脏线粒体质量相关蛋白表达,促进肝细胞更新、重塑。基于UPRmt在肝脏组织中的作用研究发现,补充烟酰胺腺嘌呤二核苷酸可激活肝脏UPRmt,显著降低脂肪肝的发生(Garianik et al.,2016)。相反,过表达UPRmt关键线粒体蛋白酶ClpP(Sen et al.,2019)或基因敲除ClpP(Bhaskaran et al.,2018)均能够上调UPRmt作用,改善衰老肝细胞代谢,抑制高脂膳食诱导的脂肪肝及IR现象,体现出线粒体毒性兴奋效应。此外,Yang等(2020)近期报道,miR‐29a可抑制GSK3β,降低SIRT1介导的线粒体生物发生,缓解线粒体蛋白应激与UPRmt水平,是治疗NAFLD的靶治疗因子。由此可见,UPRmt在调节肝细胞代谢中亦发挥重要作用,深入研究运动对肝组织UPRmt的调节机制将有助于进一步揭示线粒体蛋白稳态在运动干预NAFLD发生发展中的生物学机制。
3.3 运动调节UPRmt水平,促进线粒体因子分泌
虽然目前有关运动调节肝脏组织UPRmt的研究较少,更多关注运动缓解肝脏内质网未折叠蛋白反应(endo‐plasmic reticulum unfolded protein response,UPRer)(Este‐banez et al.,2018;Zou et al.,2020)及提高线粒体功能的作用(Goncalves et al.,2013,2014;Stevanovic et al.,2020),但随着研究不断深入,运动对肝脏线粒体功能及UPRmt的调节作用越来越受到关注。由于线粒体因子分泌的调节依赖于激活elF2α磷酸化及其下游ATF4‐CHOP轴,即UP‐Rmt机制。由此推测,运动促进线粒体因子释放与UP‐Rmt、线粒体毒性兴奋效应密切相关。近年来,随着对FGF21和GDF15等线粒体因子的研究逐步深入,多组织间“对话”逐渐清晰,运动可促进诸多肝脏线粒体因子分泌,参与糖脂代谢调节(Seo et al.,2021)。这些因子被广泛应用于肝脏糖脂代谢及心血管疾病的预防与康复研究,有望成为有效干预靶点之一。运动干预、食物限制、服用二甲双胍等手段对刺激线粒体因子分泌作用显著,大量围绕运动调节线粒体因子分泌的研究陆续展开(Conte et al.,2020;Geng et al.,2019;Gonzalez‐Gil et al.,2020;Klaus et al.,2021)。目前研究大多聚焦不同运动方式刺激后线粒体因子分泌的来源及水平变化情况,研究发现,肌肉在进行亚极量运动或最大离心收缩时,肝脏组织是循环血液中FGF21的主要来源,而非骨骼肌组织,且骨骼肌FGF21蛋白含量没有发生变化(Hansen et al.,2015;Parmar et al.,2018)。同样发现一次性急性运动可显著提高肝脏释放GDF15因子(Kleinert et al.,2018)。也有研究报道运动诱导线粒体因子分泌参与代谢调节的机制,例如,运动可提高肥胖个体脂肪组织成纤维细胞生长因子受体1(fibroblast growth factor receptor 1,FGFR1)及其辅受体(klotho beta,KLB)表达水平,增加FGF21敏感性,降低血液循环中FFAs含量与炎症反应,增强脂肪酸氧化水平,减少肝脏、骨骼肌中脂肪异位沉积(Lewis et al.,2019)。Gao等(2020)研究发现,运动伴随的骨骼肌收缩可促进FGF21分泌,其调节肝脏AMPK介导的脂滴自噬作用,是改善高脂膳食导致肝脏脂质代谢异常的重要机制。
4 结论
综上所述,氧化应激是NAFLD发生发展的核心环节,NAFLD发生发展中,较低ROS水平可激活UPRmt,使线粒体产生毒性兴奋效应,此时期为线粒体稳态自我修复的关键期。相反,若无外源性干预,肝细胞氧化应激持续加剧,过高水平ROS反而使UPRmt作用消退,导致线粒体稳态失衡,NAFLD发展进一步恶化。运动干预通过改善肝脏氧化应激、增强线粒体-细胞核“对话”及促进线粒体因子分泌等途径,调节UPRmt水平,激活线粒体毒性兴奋效应,在逆转NAFLD发生发展中发挥重要调控作用(图4)。
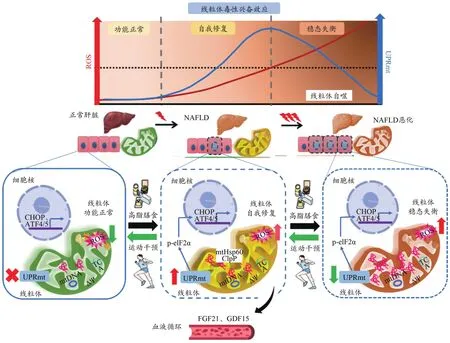
图4 运动调节UPRmt逆转NAFLD的分子机制假设图Figure 4.Molecular Mechanism Hypothesis of Exercise Regulates UPRmt and Reverses NAFLD
