分子印迹聚合物结合高效液相色谱法测定人体尿液中的8-羟基脱氧鸟苷和1-甲基鸟苷
2022-09-30吴萍萍肖慧琳宋晓丹黄丽英
吴萍萍, 肖慧琳, 宋晓丹, 黄丽英*
(1.福建医科大学药学院,福建福州 350122)(2.福建省立医院,福建福州 350001)
结直肠癌(colorectal,CRC)是常见的消化道恶性肿瘤之一,在我国的发病率呈逐年上升的趋势[1],寻找新颖的肿瘤标志物对恶性肿瘤的早期预警具有十分重要的意义。尿液中的修饰核苷排放量可反映机体中tRNA的降解速率,进而确定机体内细胞增殖的情况。当肿瘤患者体内的肿瘤细胞迅速增长时会加快tRNA的合成速度,尿液中的修饰核苷的排放量也随之相应升高[2]。因此尿中修饰核苷的含量测定,对于肿瘤的早期诊断、治疗和预后情况跟踪具有重要的临床意义。研究发现[3 - 5],结直肠癌患者血清、组织和尿液中8-羟基脱氧鸟苷(8-OHdG,8-hydroxy-2-deoxyguanosine)较正常对照组显著增加,8-OHdG水平升高是早期结肠癌与结直肠腺瘤的危险因素。冯波等[6]对结直肠癌患者尿液中的14种修饰核苷进行分析,发现假尿苷和1-甲基鸟苷(M1G,1-methylguanosine)具有较高敏感性,且与肿瘤大小与病理分期相关,更具临床应用前景。
目前,尿中8-OHdG和M1G的检测方法主要包括生物检测法和色谱法等,其中色谱法是现在最普遍的方法。生物检测法主要是酶联免疫吸附法(ELISA)[3],该方法操作简单,重现好,但是抗体间容易存在交叉反应,可能导致检测值相较于真实值偏高。色谱分析法主要包括高效液相色谱法(HPLC)[7]、高效液相色谱串联质谱法(HPLC-MS)[8]和超高效液相色谱串联质谱法(UPLC-MS)[9]。虽然HPLC-MS和UPLC-MS具有较高灵敏度和选择性,但是存在仪器成本和技术要求较高等缺点,限制了其广泛应用。
人体尿液中化学成分十分复杂,修饰核苷含量较低且种类多,采用HPLC法检测前往往需要进行预处理。分子印迹聚合物(Molecular Imprinting Polymers,MIPs)是一种人工合成的高分子材料,具有高特异性、性质稳定、制备简单和成本低廉等优点[10]。将其作为分散固相微萃取(Dispersive Solid Phase Extraction,DSPE)的吸附剂,能够更好地除去基质干扰,选择性地从复杂的样品中分离和富集目标分析物,有效提高分析方法的准确度和灵敏度。本次研究以8-羟基脱氧鸟苷分子印迹聚合物作为分散固相微萃取吸附剂,将其用于结直肠癌患者尿液中的8-OHdG和M1G分离与富集,并结合HPLC对其含量进行测定。
1 实验部分
1.1 仪器与试剂
LC-15C高效液相色谱仪(日本岛津公司);Agilent TC-C18色谱柱(4.6×250 mm,5 μm)(美国安捷伦科技有限公司);BS124S电子天平(北京赛多利斯仪器系统有限公司);SZ-93自动双重水蒸馏器(上海亚荣生化仪器厂);IKA Vortex Genius 3涡旋振荡器(德国艾卡集团);KQ-250DE型数控超声波清洗器(昆山市超声仪器有限公司);FD-1-50冻干机(北京博医康实验仪器有限公司);eppendorf 5430r高速冷冻离心机(德国艾本德股份公司)。
肌酐(标准品,99%)、乙二醇二甲基丙烯酸酯(EGDMA)(98%)、十二烷醇(99%)和2,2-偶二氮二异丁腈(AIBN)购自阿拉丁试剂有限公司;甲基丙烯酸(MAA)(99%)购自damas-beta;二甲基亚砜(DMSO)购自上海沪试化工有限公司、1-甲基鸟苷(标准品,98%)和8-羟基脱氧鸟苷(标准品,95%)购自上海源叶生物科技有限公司;乙酸铵和NaCl购自西陇科学股份有限公司;甲醇(色谱级)购自Sigma-Aldrich试剂公司;除特别说明外,其它试剂均为分析纯,实验室用水为二次蒸馏水。
1.2 标准品工作液配制
肌酐标准储备溶液:精密称取0.100 g肌酐标准品粉末,用水定容至10 mL容量瓶(10.00 mg/mL),置于4 ℃保存备用,根据需要将储备液用水稀释到一定浓度的标准品工作液。1-甲基鸟苷标准储备溶液:精密称取0.0100 g 1-甲基鸟苷标准品粉末,用水定容至10 mL容量瓶(1.000 mg/mL)置于-20 ℃冷冻保存备用。8-羟基脱氧鸟苷标准储备溶液:精密称取0.0100 g 8-羟基脱氧鸟苷标准品粉末,用水定容至10 mL容量瓶(1.000 mg/mL)置于-20 ℃冷冻保存备用。使用时根据需要将8-OHdG和M1G两种标准品储备液用水稀释至一定溶度的混合标准品工作液。
1.3 液相色谱分析条件
色谱柱:Agilent TC-C18(4.6×250 mm,5 μm);流动相:A为0.03% 乙酸-5 mmol/L乙酸铵,B为甲醇。梯度洗脱程序为:0~5 min,2%B;5~10 min,2%~8%B;10~35 min,8%B;35~40 min,8%~2%B。流速:1.0 mL/min;检测波长:254 nm;柱温:30 ℃,进样量:20 μL。
1.4 分子印迹聚合物的制备
实验参考文献报道方法[11,12],采用本体聚合法制备。在50 mL的EP管中加入15 mL的DMSO和12 mL十二烷醇溶液,加入模板分子8-羟基脱氧鸟苷(0.5 mmol)、功能单体MAA(2 mmol)超声混匀孵育15 min,形成模板分子-功能单体复合物。随后加入交联剂EGDMA(10 mmol),超声混匀后加入引发剂AIBN(30 mg),超声脱气5 min,通氮气5 min除氧,密封,放入60 ℃恒温水浴锅中恒温聚合24 h,得到块状固体聚合物。经研磨后,加入甲醇-乙酸(90∶10,V/V)涡旋洗脱模板分子,7 830 r/min下离心2 min,弃去洗脱液,重复多次,直至洗脱液在高效液相254 nm处无检测到模板分子对应的色谱峰。真空干燥至恒重,得到白色粉末(MIPs)研磨过200目筛备用。除不加模板分子8-羟基脱氧鸟苷外,按照同上步骤制备非分子印迹聚合物(NIPs)。
1.5 MIPs的吸附性能测试
1.5.1 MIPs吸附动力学的测定精密称取10.0 mg已制备好的MIPs,分别置于50 mL EP管中,各加入5 mL 50 μg/mL 8-羟基脱氧鸟苷水溶液,于20 ℃下恒温涡旋0、1、2、3、4、5、6、7 min后,7 830 r/min离心5 min。上清液经微孔滤膜过滤后,用HPLC分别测定上清液中8-羟基脱氧鸟苷水溶液的浓度,平行测定3次取平均值,利用差减法计算吸附量Q(mg/g)随时间t(min)的变化值[13]。
1.5.2 选择竞争性实验本实验选择与8-羟基脱氧鸟苷(底物)分子结构相似的3种核苷类化合物1-甲基鸟苷、腺苷和肌苷为竟争底物进行比较,考察MIPs和NIPs对竟争底物和底物的选择性吸附能力与分子识别性能。
比较单独和分别与8-羟基脱氧鸟苷混合后在MIPs上的吸附情况,计算不同竟争底物和底物在MIPs和NIPs上的吸附量Q(mg/g),采用静态分配系数KD、分离因子α和相对分离因子α′来表征聚合物对竟争底物的选择性吸附能力。KD、α和α′的计算公式参照文献方法[14]。cs和s分别表示竟争底物及底物,当KDcs=KDs时,α=1。KD越高,表示聚合物对该竞争底物或底物的结合量越大,对其特异性识别能力超强;α值越高,表示聚合物对底物的选择性越好;α′越高,表示印迹聚合物MIPs的选择性比非印迹聚合物NIPs越好。
1.6 样品收集与处理
疾病组:结直肠癌患者8名,所有患者均在超声定位下行穿刺活检确诊,随机采集尿样。对照组:健康志愿者8名,随机采集尿样。尿样保存:尿样收集后立即置于-20 ℃密封保存,备用。
冻存的尿样于室温解冻,7 830 r/min下离心10 min,取上清尿样用水定容至50 mL容量瓶,取10 mL作为萃取供相备用,调节pH=7.00,加入30.0 mg MIPs,于10 ℃下涡旋1 min,7 830 r/min下离心5 min,弃去上清液,加入1 mL1%乙酸水溶液,涡旋30 s,重复三次,合并3次上清液,于-20 ℃冷冻过夜,在-50 ℃,40.0 Pa下冷冻干燥10 h,加入50 μL双蒸水,12 000 r/min下离心10 min后,取上清液20 μL进样分析。
2 结果与讨论
2.1 MIPs和NIPs的表征
2.1.1 MIPs和NIPs的电镜表征图1分别为MIPs和NIPs的扫描电镜(SEM)图,由图1(A)可见MIPs聚合材料表面较为粗糙,具有更多孔穴;由图1(B)显示,NIPs聚合材料表面更为光滑些,孔穴较少。

图1 MIPs(A)和NIPs(B)的扫描电镜(SEM)图Fig.1 SEM images of MIPs(A) and NIPs(B)
2.1.2 MIPs和NIPs的红外光谱表征图2为未经过洗脱的MIPs(a)、洗脱后的MIPs(b)及NIPs(c)的红外光谱图。在图2a中8-羟基脱氧鸟苷在1 540 cm-1处有一个C=N的特征吸收峰,证明模板分子已经成功印迹到MIPs中,而图2b和图2c几乎没有太大区别,说明洗脱过后,MIPs中的模板分子已经去除干净。由图2显示,3 500~3 400 cm-1是O-H和N-H的伸缩振动峰,1 731cm-1是C=O的伸缩振动峰,1 642 cm-1是C=C的伸缩振动峰,1 266 cm-1是羧基中C-O 的伸缩振动峰,证明了聚合物中功能单体MAA的存在,1 144cm-1是C-O-C的伸缩振动峰,证明了聚合物是由EGDMA合成的。
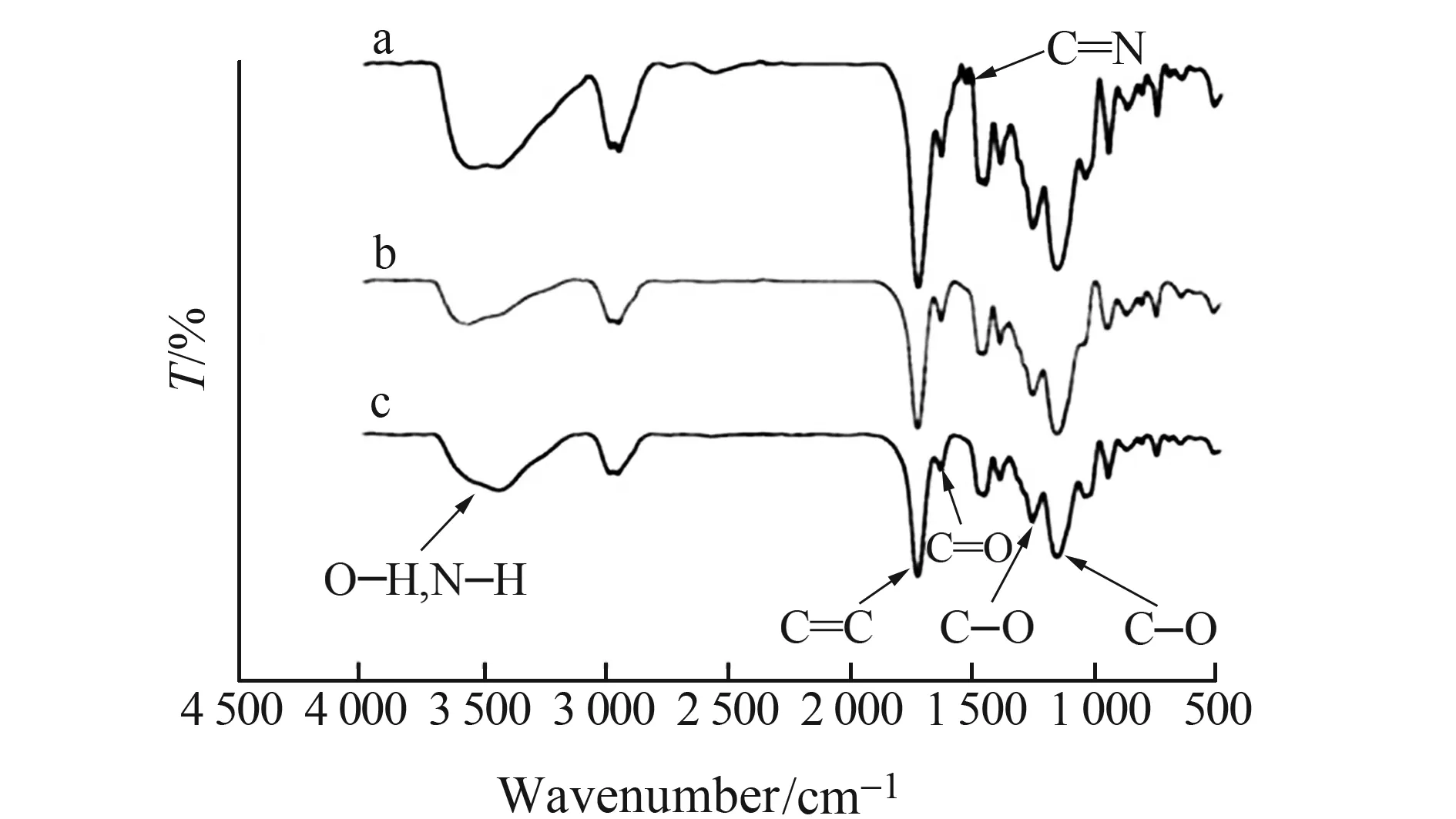
图2 分子聚合物的红外光谱图a.未经洗脱过MIPs;b.洗脱后的MIPs;c.NIPsFig.2 Infrared spectra of MIPs and NIPsa:MIPs before of extraction;b:MIPs after of extraction;c:NIPs.
2.2 动态吸附曲线
吸附过程的动力学研究可用来描述吸附剂吸附溶质的速率快慢,将数据通过动力学模型进行拟合,从而探讨该吸附剂的吸附机理。根据“1.5.1”方法得到由7个时间点和对应的吸附量(Q,mg/g)的MIPs吸附动力学曲线(图3)。由图3可知,在0~1 min,吸附量迅速增大,说明该阶段吸附速度很快,2~4 min,吸附量随时间继续增大,但是吸附速度明显下降,4 min之后,吸附量基本不变,吸附动力学曲线趋于平衡,说明吸附剂对8-羟基脱氧鸟苷的吸附达到饱和。
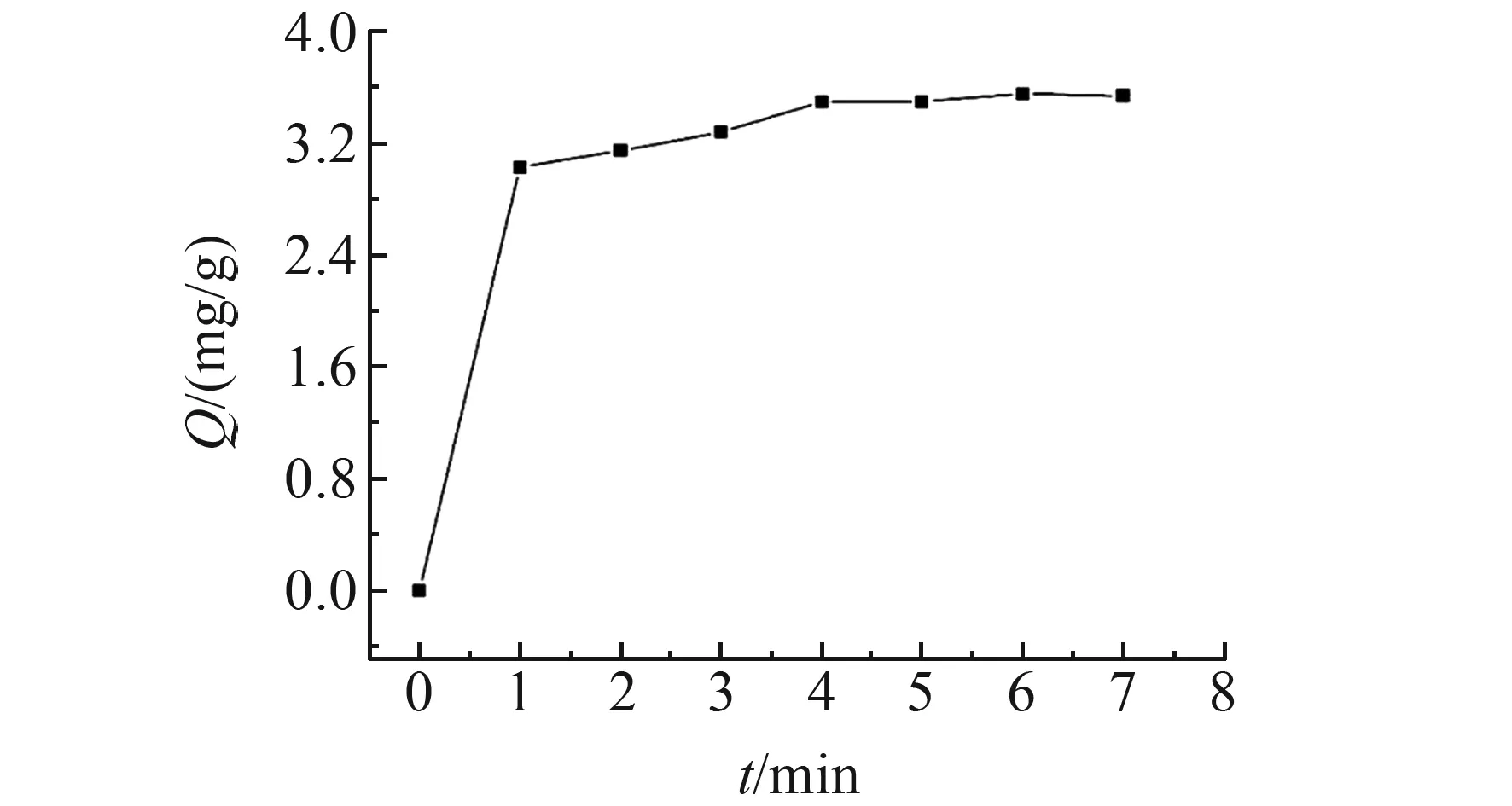
图3 MIPs的吸附动力学曲线图Fig.3 Adsorption kinetics curve of MIPs
2.3 选择竞争性实验
为了评价吸附剂对8-羟基脱氧鸟苷的选择性实验及干扰物对目标物吸附的影响,需绘制目标物和干扰物的峰面积A对c(μg/mL)的标准曲线,以求得各物质吸附后的浓度。腺苷:A=45844c+9716(r2=1);1-甲基鸟苷:A=102995c-45198(r2=0.9999);肌苷:A=52126c+8235.2(r2=0.9999);8-羟基脱氧鸟苷:A=56095c-3815(r2=1)。
用吸附等温曲线分别测定MIPs和NIPs对水中目标分子和干扰物的吸附量,计算分子印迹聚合物对不同干扰物的静态吸附量Q、对底物的结合分配系数KD、分离因子α和相对分离因子α′,从而比较MIPs对目标分子和干扰物的选择性,结果见表1。
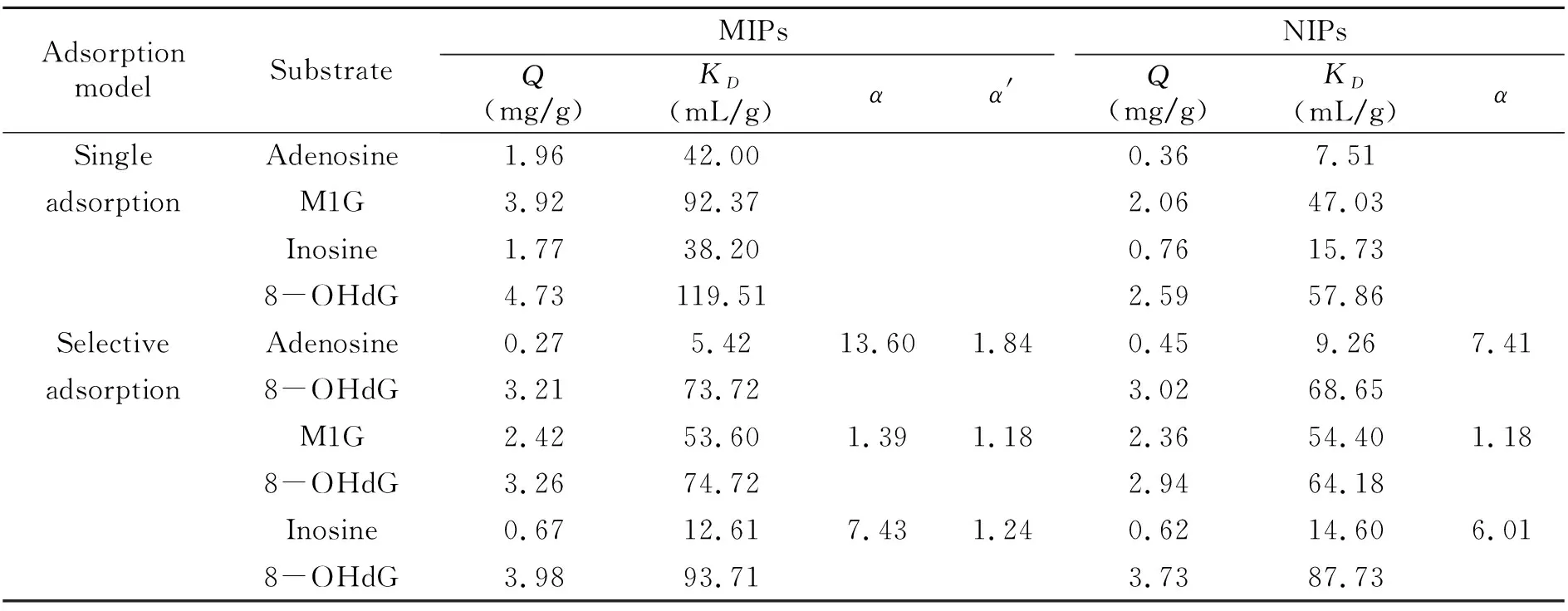
表1 MIPs和NIPs对不同目标物选择性吸附实验
由表1可知,单独吸附时,MIPs对底物腺苷、1-甲基鸟苷、肌苷和8-羟基脱氧鸟苷的吸附量均高NIPs对底物的吸附量,表明MIPs对8-羟基脱氧鸟苷及其类似物有一定的印迹效应。MIPs对8-羟基脱氧鸟苷的分配系数KD最高,其次是1-甲基鸟苷。在选择性吸附实验中,可知MIPs对8-羟基脱氧鸟苷与其他底物的分离因子α均大于1,表明MIPs能够较好的分离8-羟基脱氧鸟苷和其他结构相似的底物,α值越高,说明越容易分离。α′大于1表明了MIPs对8-羟基脱氧鸟苷和底物的相对分离能力强于NIPs。综上各项参数所述,MIPs和NIPs在空间上存在差异,MIPs对8-羟基脱氧鸟苷具有更好的吸附性和选择性。
2.4 分散固相萃取条件的优化
分散固相萃取过程中,使用涡旋振荡器的目的是使MIPs与混合目标物充分接触并将其吸附。实验研究了不同的涡旋时间,即15、30、60、120、180、240和300 s的吸附效果,在60 s时对两个目标物的吸附量达到峰值,但是继续涡旋吸附量会降低,可能是涡旋产热,影响了吸附。实验研究了不同洗脱溶剂对洗脱率的影响。对于MIPs而言,酸性条件有利于减弱氢键的作用,因此采用不同浓度的酸溶液进行实验。分别选择了乙酸水溶液(0.1%、0.5%、1.0%、2.0%和3.0%),甲酸水溶液(0.1%、0.5%和1.0%)作为洗脱剂溶液。如图4所示,1%乙酸溶液的洗脱效果最好,继续增大乙酸的浓度反而降低洗脱率。考虑到8-OHdG 和M1G的极性较大,所以本实验选择1%乙酸-乙腈溶液、1%乙酸-甲醇溶液以及正丁醇饱和的1%乙酸水溶液作为洗脱液进行试验。结果表明,正丁醇饱和的1%乙酸水溶液和1%乙酸-甲醇溶液对8-OHdG和M1G的洗脱效果不理想且影响两个目标物的峰型,1%乙酸-乙腈水溶液洗脱效果很差,几乎不能洗脱目标分子,1%乙酸水溶液洗脱效果最好,因此选择1%乙酸水溶液作为洗脱溶剂。
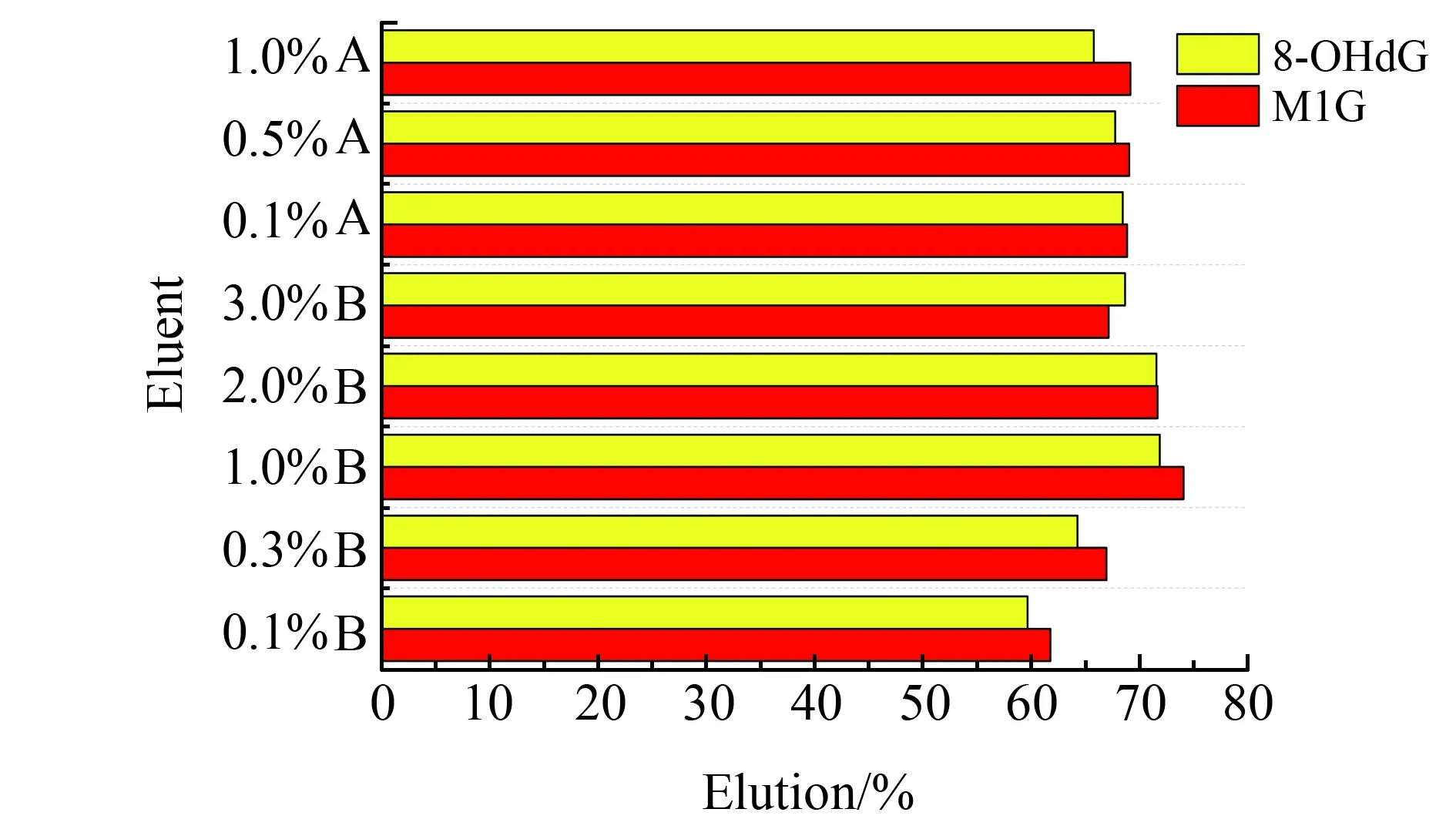
图4 洗脱溶剂的选择Fig.4 The optimization of eluentEluent:A,Formic acid;B.Acetic acid.
3 方法学考察
3.1 定量标准曲线及方法检出限
配制浓度范围为10~1 000 ng/mL 8-OHdG和20~2 000 ng/mL M1G系列的混合标准工作液,在最佳条件下进行萃取后结合高效液相色谱法进样分析。分别以萃取前标准品溶液浓度为横坐标(X,ng/mL)以及萃取后的峰面积(Y)为纵坐标,进行线性回归,得工作曲线。肌酐标准品溶液不经过萃取,以峰面积(Y)为纵坐标,标准品浓度(X,μg/mL)为横坐标,进行线性回归得到工作曲线。结果表明,3个物质在各自线性范围内的线性关系良好,3个曲线的相关系数r2≥0.9996。萃取后的回归方程、相关系数(r2),线性范围(ng/mL)、检测限和定量限见表2。
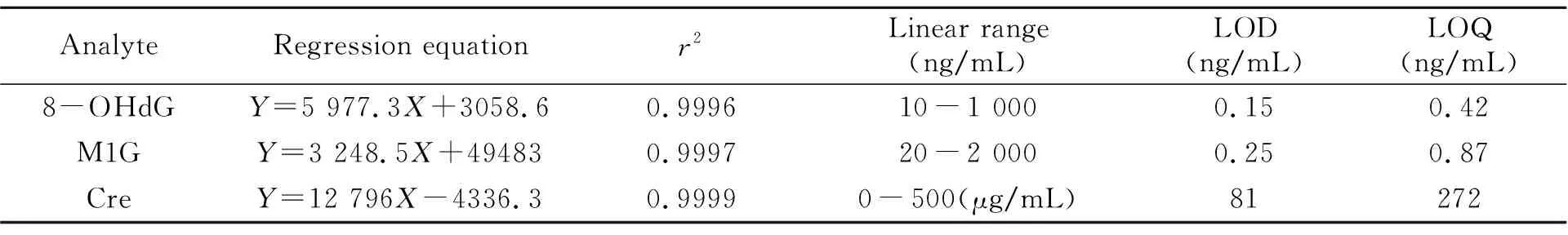
表2 修饰核苷的回归方程、相关系数(r2)、线性范围、检测限、定量限
3.2 精密度实验
选取50 ng/mL的含有2种目标修饰核苷(8-OHdG和M1G)的混合标准溶液,在最佳条件下进行萃取,进样分析(每日平行测定6次,连续测定3天)。选取50 μg/mL的肌酐标准品溶液直接进样分析。测得日内精密度的相对标准偏差(RSD)小于3.2%,日间精密度RSD小于5.8%。
3.3 回收率实验
取正常对照组尿液3份,按高、中、低3个水平分别精密加入适量的混合标准品溶液(8-OHdG、M1G和Cre)并稀释至50 mL,每个添加水平平行测定3次,在最佳条件下萃取2个目标目标物。回收率实验结果见表3,该方法在尿样中的加标回收率在82.6%~93.2%之间。
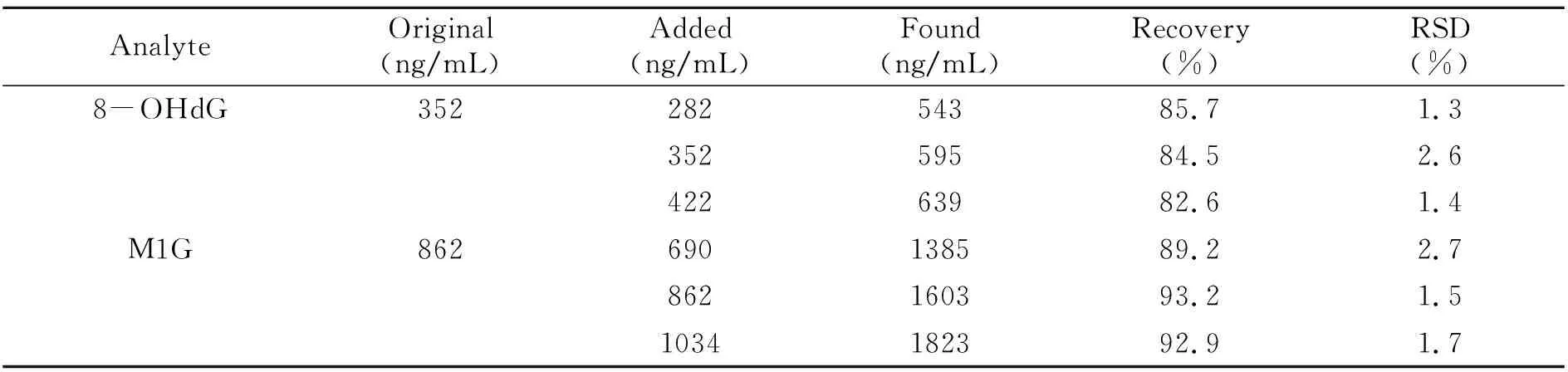
表3 尿液中两种修饰核苷的回收率和相对标准偏差(n=3)
3.4 实际样品分析
图5A为浓度1 000 ng/mL的混合标准品8-OHdG和M1G分别经NIPs和MIPs处理后得到的色谱图,可以看出8-OHdG和M1G均具有很好的富集效果,其中萃取前Cre、M1G和8-OHdG的保留时间分别为4.646、26.428、35.515 min,萃取后的M1G和8-OHdG的保留时间为26.304、35.238 min。
应用实验合成的8-OHdG-MIPs,结合HPLC法对健康对照组和结肠癌疾病组尿样进行分析测定,每个样品平行3次重复测定,取均值为含量值。尿样萃取前和萃取后对比色谱图如图5B所示。尿中修饰核苷在一天中的不同时刻的排放量不尽相同,即其排放量变化差异大,但尿中修饰核苷与肌酐(Creatinine,Cre)的比值却是恒定的[15],且受食物影响很小[2]。因此,采用修饰核苷和肌酐的比值(nmol/μmol)来反映正常人和结直肠癌患者尿液中的修饰核苷的含量。肌酐的浓度可直接由萃取前尿样测得。实验分别对8名健康志愿者和结肠癌患者的尿样(sampleA)进行测定,8-OHdG/Cre(nmol/μmol)和M1G/Cre(nmol/μmol)的比值结果如表4所示,结肠癌患者尿样中的8-OHdG/Cre(P=0.02)和M1G/Cre(P=0.00003)的含量比值水平显著高于健康志愿者(P<0.05),差异具有统计学意义。
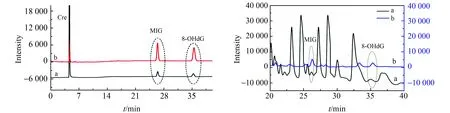
图5 萃取前后对比图Fig.5 HPLC chromatograms obtained from mixed standard solutions and urine sample before and after extractionA-a the mixed standard solutions before extraction;A-b the mixed standard solutions after extraction.B-a the urine sample of Colon cancer patients before extraction;B-b the urine sample of Colon cancer patients after extraction.
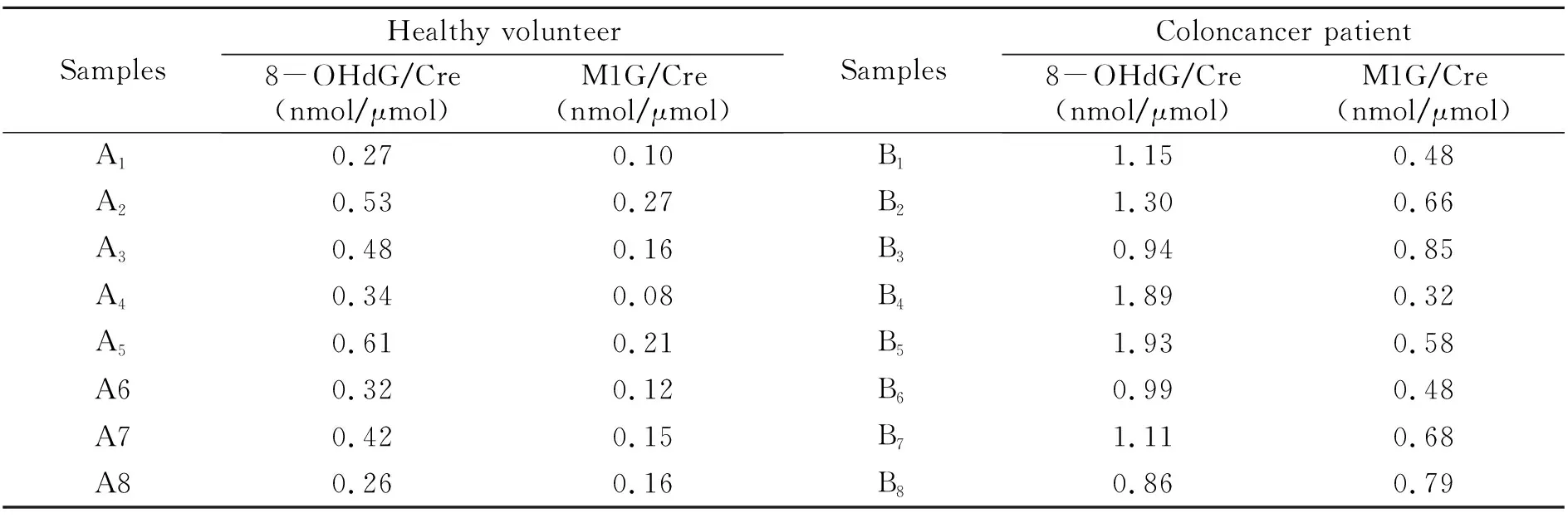
表4 尿液样品中修饰核苷/肌酐比值(n=3)
4 结论
成功制备了8-羟基脱氧鸟苷分子印迹聚合物,其表现出较好的特异性吸附,将其作为分散固相萃取吸附剂,结合高效液相色谱法测定结肠癌患者尿液中的8-OHdG和M1G。该方法能够有效地去除尿液中复杂基质的干扰,提高分析的准确性,为尿液中的8-OHdG和M1G作为结直肠癌肿瘤标志物的前处理和分析提供了新的思路,使尿中修饰核苷检测用于临床成为可能。
