LHCGR基因复合杂合突变导致小阴茎1例
2022-09-27杨宇帆赵亚玲伍学焱茅江峰
杨宇帆,赵亚玲,王 曦,聂 敏,伍学焱,茅江峰
(中国医学科学院 北京协和医学院 北京协和医院 内分泌科, 北京 100730)
黄体生成素(luteinizing hormone, LH)或人绒毛膜促性腺激素(human chorionic gonadotropin, HCG),能够结合促黄体生成素/绒毛膜促性腺激素受体(luteinizing hormone/choriogonadotropin recep-tor,LHCGR)而产生睾酮。LHCGR蛋白属于经典的7跨膜G蛋白偶联受体。当配体和受体结合后,受体的构象发生变化并激活G蛋白。通过腺苷酸环化酶-环磷酸腺苷(cAMP)-蛋白激酶A(PKA)信号传导途径,促进雄激素合成[1]。
睾酮在促进男性性别分化和生殖成熟方面发挥重要作用[2]。在早期胚胎发育过程中,来源于胎盘的HCG与胎儿睾丸间质细胞(Leydig)中的LHCGR蛋白结合,促进间质细胞成熟并产生睾酮[3]。在青春期前后,睾酮可促进并维持男性第二性征,促进精子生成[4]。
LHCGR基因位于染色体2p21上,包含11个外显子。与其他G蛋白偶联受体相似,LHCGR蛋白含有一个大的细胞外结构域,可与配体结合。它含有7个跨膜螺旋进行信号传导[4]。LHCGR蛋白由699个氨基酸组成,主要在男性的间质细胞膜和女性颗粒细胞和黄体中表达[5]。
LHCGR基因突变可导致各种疾病发生。男性LHCGR激活突变可导致非LH依赖性性早熟和Leydig细胞瘤。失活突变则导致睾丸间质细胞发育不良和睾酮合成障碍,表现为小阴茎和/或尿道下裂或不育。在女性,LHCGR失活突变导致月经稀发、闭经和不孕症[5]。
本文报道1例高促性腺激素性性腺功能减退症的患者,因LHCGR基因发生复合杂合突变导致小阴茎。虽然临床表现较为单一,但有助于增加对致病基因和临床表现的认识。
1 材料与方法
1.1 研究对象
北京协和医院内分泌科收治的1例12岁小阴茎男童。根据病史、生殖器特征和实验室检查结果,诊断为“高促性腺激素性性腺功能减退症”。经患者和其父母知情同意,抽取外周血,提取基因组DNA进行二代测序panel分析。本研究通过北京协和医院伦理委员会审批(编号JS-2111)。
1.2 研究方法
1.2.1 实验室化验和影像学检查:实验室检查和影像学检查分别由北京协和医院检验科和放射科完成。
1.2.2 基因组DNA提取:按照操作说明书,提取患者及父母外周血白细胞DNA,进行二代测序panel。
1.2.3 测序:按照操作说明书,使用高通量测序系统Illumina Nextseq 500对产物进行测序。
1.2.4 测序结果分析:二代测序的定制化基因panel检测结果,参考的基因组版本为GRCh37/hg19,并参考千人基因组、ESP6500(NHLBI Exome Sequencing Project)、EXAC(The Exome Aggregation Consortium)和EXAC-EAS(EXAC约4000个东亚人)的数据库。将致病基因的测序结果与NCBI网站数据库进行比对分析(GenBank编号LHCGR),并与人类基因突变数据库(Human Gene Mutation Database,HGMD)进行比对。
1.2.5 突变验证: 针对检测出的变异位点设计特异引物,进行Sanger测序,采用 ABI3730xl DNA 测序仪进行双端测序。针对c.233+1G>A突变,正向引物为5′-GCATTCAGTCATTTGAAGCAAG-3′,反向引物为5′-TCCTCAGCCTGAATCCAGTTC-3′;针对c.547G>A突变,正向引物为5′-GTTTCTAGCCAGC CAGTTGC-3′,反向引物为5′-ATTGATGGTGGTGGT GATGA-3′。
2 结果
2.1 临床资料
患儿出生时为男性外阴,阴茎长1 cm,阴囊发育差,双侧隐睾。1岁行双侧隐睾下拉固定术。随着年龄增加,阴茎略增长,无阴毛增长。目前患儿12岁,阴茎长度3 cm(图1A),嗅觉正常。父母非近亲婚配,否认类似遗传病史。父亲青春期发育正常,身高168 cm。母亲青春期发育正常,身高162 cm,G1P1,月经规律。
2.2 体格检查
腹型肥胖,身高162 cm,体质量58 kg。阴茎长度3 cm,周径4 cm,阴毛P1。双侧腹股沟陈旧手术疤痕,双侧睾丸体积1 mL(图1A)。乳房B1。
2.3 辅助检查
LH 31 IU/L↑(N 0.5~6.5 IU/L),FSH 35 IU/L↑(N 0.4~5.3 IU/L),雌二醇 44 pg/mL(N 35~65 pg/mL),睾酮0.99 nmol/L↓(8.4~12.9 nmol/L)。ACTH 36 pg/mL(N 5~65 pg/mL),血总皮质醇(8am) 176 nmol/L (N 133~537 nmol/L)。其他生化检查和血尿常规未见异常。
2.4 诊治和随访
根据临床表现和生化检测,诊断“高促性腺激素性性腺功能减退症”。针对小阴茎,给予口服十一酸睾酮(胶囊),每日两次,每次40 mg。用药12个月后复诊,阴茎长度从1 cm 增加到5 cm,阴毛P3。
2.5 突变位点功能预测
测序结果显示,LHCGR基因存在复合杂合突变。一个突变为c.233+1G>A,位于第2外显子和第2内含子交界处,来源于母亲(图1B)。该突变会引起mRNA剪切异常。文献数据库中没有该位点的报道。根据美国医学遗传学与基因组学学会(ACMG)指南[6],此剪切突变为低频突变,符合PVS1+PM2,判定为疑似致病性变异。
Splice Site Score Calculation软件预测,该突变导致剪切分数为-1.5,远小于正常值8.1,提示该位点无法正常剪切。由于第2外显子不能被完整剪接出来,导致第2外显子的碱基数量过长,翻译后形成的氨基酸数量增多,形成69个氨基酸(VIKM*VSYCISKDIRNWNNEFFLRENTFERG*FF*ERNCV*DTHIFLSFEISQIDSLERIEANAFDNLL),比正常的第2外显子(含有24个氨基酸,VVPPGGKGAGPLFYTLSSHWMRAA)多了45个氨基酸。与此相应,三维结构蛋白质模型也显示,突变后的蛋白结构和正常受体结构相比,差异甚大(图2)。
另一个杂合突变为c.547G>A,位于第7外显子,来源于父亲(图1C)。该突变导致第183位的甘氨酸变为精氨酸(p.G183R)。文献数据库中没有该位点的报道。根据美国医学遗传学与基因组学学会(ACMG)指南[6],此低频突变,符合PM2+PM3+BP4,判定为“临床意义未明”。功能预测软件PolyPhen、Mutation Taster、PANTHER提示突变为良性、致病性和可能致病。模拟的蛋白质三维结构显示,突变LHCGR受体蛋白氢键增多,可能影响蛋白极性(图3)。突变的氨基酸位于受体的胞外区域,可能干扰与配体的结合。
未检测到其他和先天性低促性腺激素性性腺功能减退症或性发育异常相关的基因突变。
3 讨论
本例患儿以“小阴茎,低睾酮,高LH”为突出表现,基因检测证实LHCGR基因存在剪接位点突变(c.233+1G>A)和错义突变(c.547G>A)。因此, 临床诊断LHCGR基因失活突变致小阴茎基本明确。

A.micropenis and old surgical scar of groin; B.a heterozygous mutation c.233+1G→A occurred at the splicing site of the LHCGR gene (between exon 2 and intron 2), with the upper, middle and lower base sequences representing the patient, father and mother, respectively; the arrow indicated the base mutation c.233+1G→A, which originated from the patient’s mother; C.the LHCGR gene is subjected to a heterozygous mutation c.547G→A in exon 7, with the upper, middle, lower base sequences representing the patient, father and mother, respectively; the c.547G→A mutation was derived from the patient’ s father

A.prediction model of normal LHCGR receptor protein; B.Due to the c.233+1G>A splicing anomaly, a prediction model of a completely different protein was formed图2 正常和c.233+1G>A突变的LHCGR蛋白三维预测模型Fig 2 Prediction 3D models of normal and mutant(c.233+1G>A) LHCGR receptor protein
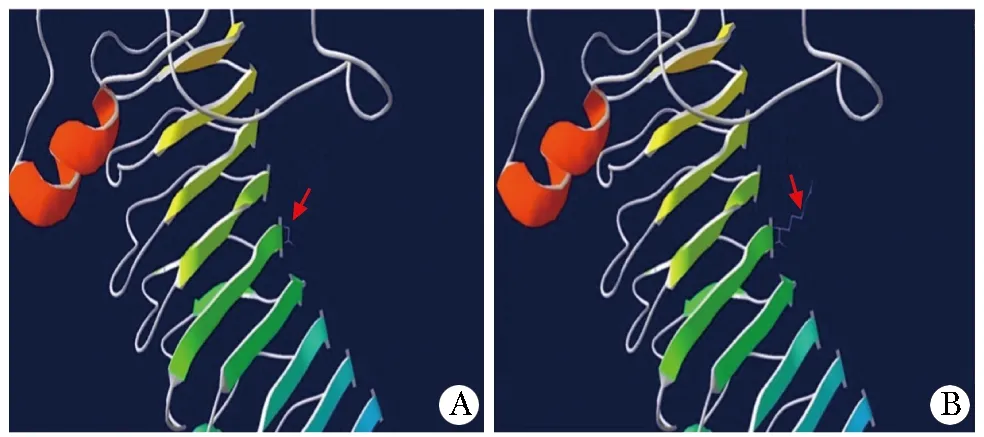
A.three-dimensiona protein prediction model of normal LHCGR receptor; the red arrow indicates normal hydrogen bond; B.a three-dimensional protein prediction model of G183R mutant LHCGR; the red arrow shows an increase in hydrogen bonds, which may lead to abnormal protein function图3 正常和c.547G→A突变的LHCGR受体蛋白三维结构预测模型Fig 3 Prediction 3D models of normal and mutant(c.547G>A) LHCGR receptor protein
间质细胞发育不良(Leydig cell hypoplasia,OMIM #238320)是引起男性性分化异常的罕见病因。黄体生成激素/绒毛膜促性腺激素受体(LHCGR)基因的失活突变可导致间质细胞发育不良。有文献描述了因1例LHCGR复合杂合突变导致的性分化异常男性患者,基因突变包括剪接位点突变(c.681-1 G>A)和移码变异体(c.1582_1585del ATAT,p.Ile528),临床表现为睾酮合成障碍和女性外阴[7]。而本例患儿仅表现为小阴茎,可能和患者基因突变导致的功能损伤较轻有关。
本例患者LHCGR的1个基因突变为c.547G>A(p.G183R)。多个功能预测软件提示为致病性突变,模拟的三维结构显示突变型的LHCGR受体蛋白氢键增多,可能影响蛋白的极性。有报道1例由于LHCGR基因发生纯合突变c.580T>G(F194V)性分化异常男童,体外功能研究显示,194位氨基酸的改变导致LHCGR蛋白在细胞膜上的表达减少,HCG刺激后cAMP生成降低[8]。值得注意的是,本例患儿第183位氨基酸和文献报道的第194位氨基酸位于同一功能域LRR4(配体结合功能域4),因此也可能影响蛋白功能。在LHCGR基因失活突变中,有70%的突变导致LHCGR蛋白在细胞膜表达下降。这些患者均表现为两性畸形或小阴茎[9]。
另一个突变是来自母亲的c.233+1G>A,位于第2外显子和第2内含子交界处。多种预测软件都显示,突变导致剪切异常而致病。LHCGR基因发生剪切位点突变并不少见,发生在内含子区域的有c.537-3C>A、c.681-1G>A、c.955-1G>A[7,10-11],发生在外显子区域的有c.557A>C、c.558G>C和c.580A>G[12-13]。这种类型突变的男性常表现为小阴茎,提示突变仅引起蛋白功能轻度降低[11]。这与本例患者的临床表现相似。
综上所述,本研究推测上述两个位点的复合杂合突变(图4)可能导致LH受体功能障碍,导致不能合成睾酮,引起阴茎短小或两性畸形的临床表现[5]。而患者父母均为致病基因携带者,故表现正常。

图4 患者LHCGR基因复合杂合突变示意图
在另一方面,LHCGR基因也会发生激活性突变,引起睾丸间质细胞不断自主合成睾酮,表现为家族性限男性性早熟[14],但相同的突变不会导致女性周围性性早熟[5]。
儿童期开始用雄激素治疗,能够促进阴茎增大[15-16]。但过多雄激素暴露可能导致骨龄提前而影响患儿终身高。因此,需要选择合适的剂量和疗程。本患儿应用小剂量十一酸睾酮治疗12个月后,阴茎长度明显增加,提示治疗效果良好。预计随着治疗时间延长,患儿可获得更好的第二性征发育。但由于LHCGR突变导致睾丸本身不能合成睾酮,因此产生精子的可能性小。
本研究存在一定局限性,未对突变基因进行体外功能验证。虽然软件和蛋白模型能够预测位点突变导致第2外显子不能正确的剪切,但软件预测并不能取代体外功能试验。本实验也尝试利用患者外周血mRNA以验证剪切位点突变导致第2外显子剪切异常,但反转录未能获得足量mRNA。这可能和LH受体只表达在睾丸间质细胞,而在外周血中表达太低有关。
综上所述,本研究通过描述1例LHCGR复合杂合突变导致的小阴茎病例,扩展了对LHCGR基因突变导致性发育异常发病机制的认识。同时,通过治疗反应,证实了睾酮替代对这类疾病的有效性,为该病的临床治疗提供了有益的借鉴。
