PMMA/DEAM-rGO 纳米复合材料的制备及性能
2022-09-26闫杰刘喜军翟文颖
闫杰,刘喜军,翟文颖
(齐齐哈尔大学材料科学与工程学院,黑龙江齐齐哈尔 161006)
由于聚甲基丙烯酸甲酯(PMMA)硬度高、质量轻且具有高透光性,可广泛应用于光学材料、传感器、汽车、太阳能和显示装置等行业。将不同的纳米粒子引入到PMMA 中可以增强其光学、热学、电学和阻燃性能。因此研究改性PMMA 具有广阔的应用发展前景。
石墨烯是一种由碳原子以sp2杂化轨道组成的呈六角型蜂巢晶格的新型二维碳纳米材料,由于其具有优异的导电性能、导热性能以及巨大的比表面积,成为一种至关重要的复合材料填充物[1-3],在PMMA 纳米复合材料研究领域取得重大进展[4-7]。然而,当石墨烯在PMMA 基体中团聚时,PMMA/石墨烯纳米复合材料内部会出现较大的应力集中,从而削弱其优异性能,造成这种破坏的主要原因是PMMA/石墨烯界面化学的变化。因此,研究界面性质并了解界面键合类型(如范德华力、氢键、配位键和共价键)对界面力学行为的影响对于纳米尺度的界面来说是不可或缺的[8-9],在制备性能优异的PMMA/石墨烯纳米复合材料时,不仅需要实现石墨烯在PMMA 基体中的均匀分散而且要显著改善石墨烯在PMMA 基体内的界面粘附性[10-11]。
笔者首先制备甲基丙烯酸甲酯(MMA)/甲基丙烯酸二乙胺基乙酯(DEAM)-还原氧化石墨烯(rGO)分散液,借助DEAM 的助分散作用提高rGO 与MMA 的相容性,进一步促进rGO 在MMA中的均匀分散[12-14],然后采用悬浮聚合方法制备PMMA/DEAM-rGO 纳米复合材料,并对其进行了结构表征和性能分析。DEAM 的引入不仅实现了rGO 在PMMA 基体中的均匀分散,提高了rGO 在PMMA 基体中的界面粘附性,并且在聚合过程中DEAM 能够与MMA 发生共聚合反应,不会给聚合体系引入新的小分子杂质。
1 实验部分
1.1 主要原料
MMA:分析纯,使用前需要去除阻聚剂并蒸馏精制,天津科密欧化学试剂有限公司;
DEAM、过氧化苯甲酰(BPO):分析纯,上海阿拉丁生化科技股份有限公司;
rGO:分析纯,青岛华高新能源科技有限公司;
轻质CaCO3:分析纯,河北灵寿县泽达矿产品加工有限公司;
对苯二酚:分析纯,上海迈瑞尔化学技术有限公司;
HCl:分析纯,苏州享尚益电子材料有限公司;
去离子水:自制。
1.2 主要设备与仪器
高速离心机:TG16G 型,湖南凯达科学仪器有限公司;
偏光显微镜(PLM):XPF-330C 型,上海蔡康光学仪器有限公司;
傅里叶变换红外光谱(FTIR)仪:Spectrum One型,美国PE 公司;
X 射线衍射(XRD)仪:D8 Advance 型,德国Bruker 公司;
X射线光电子能谱(XPS)仪:ESCALAB250Xi型,美国Thermo 公司;
激光显微共聚焦拉曼光谱(Raman)仪:HR800型,法国JY 公司;
透射电子显微镜(TEM):H-7650 型,日本日立公司;
扫描电子显微镜(SEM):S-3400 型,日本日立公司;
差示扫描量热(DSC)仪:DSC204F1 型,德国Netzsch 公司;
热重(TG)分析仪:TGA5500 型,美国Waters公司;
双电测四探针测试仪:RTS-5 型,广州四探针科技有限公司;
平板硫化机:L29A 型,浙江杭州恒瑞机械制造有限公司;
冲击试验机:JJ-20 型,长春智能仪器设备有限公司;
电子万能试验机:WSM-20KN 型,长春智能仪器设备有限公司;
真空干燥箱:DZF-6090 型,上海精宏实验设备有限公司;
超声波清洗器:KQ-500DB 型,昆山市超声仪器有限公司;
电子天平:ME104E 型,梅特勒-托利多仪器(上海)有限公司。
1.3 样品制备
(1) MMA/DEAM-rGO 分散液的制备。
将0.100 g rGO 粉末以及0.100 g DEAM 和10.000 g MMA 液体依次加入30 mL 样品瓶中,然后将样品瓶置入功率为600 W 的超声波清洗器中,室温下超声震荡2 h,得到MMA/DEAM-rGO 分散液[15]。
(2) PMMA/DEAM-rGO 纳米复合材料的制备。
在250 mL 三颈瓶中加入70 mL 去离子水、3.000 g 轻质CaCO3,将三颈瓶移入水浴中室温下搅拌10 min。称取0.315 g BPO 加入到10 mL MMA/DEAM-rGO 分散液中,室温下搅拌5 min,使其充分溶解,待水浴温度达到50℃时缓慢滴加MMA/DEAM-rGO 分散液(5 min 滴完)并在N2保护下快速搅拌(搅拌速度300 r/min),然后将温度升至80℃并保持4 h。称取0.100 g 对苯二酚加入三颈烧瓶中继续反应30 min,待产物冷却至室温后,用10%HCl 反复洗涤、抽滤除去杂质,最后使用去离子水将产物洗涤至中性后放入60℃真空干燥箱中干燥24 h,得到PMMA/DEAM-rGO 纳米复合材料。
按照同样的聚合条件及方法,依次制备PMMA/rGO 和PMMA 样品,其中PMMA/rGO 样品中rGO 质量分数为MMA 用量的1%。在80℃下,将PMMA/rGO 和PMMA/DEAM-rGO 样品用丙酮萃取12 h,得到的样品分别命名为PMG1 和PMG2。
1.4 性能测试与结构表征
(1)离心沉降实验。
量取10 mL MMA/DEAM-rGO 分散液移入离心管中并将离心管放入转头内,采用高速离心机对分散液进行高速离心实验,离心时间5 min,离心速度1 000~7 000 r/min,离心实验结束后,取出离心管观察分散液沉降分层情况。
(2)PLM 分析。
用MMA 稀释MMA/DEAM-rGO 分散液(稀释10 倍),吸取一滴稀释的分散液滴在载玻片上,盖上盖玻片放到载物台上,采用PLM 观察分散液的分散情况,放大倍数为100 倍。
(3)化学结构分析。
将固体样品经60℃真空烘箱干燥12 h 后与KBr 混合、研磨、压片,液体样品直接涂覆在衰减全反射(ATR)晶体上,通过FTIR 仪对样品进行分析,波数范围为500~4 000 cm-1。
将样品研磨并黏附在导电胶表面,采用XPS 仪对样品进行元素分析,真空度为5×10-7Pa,标准C1s峰(284.6 eV)为能量校正内标。
将样品研磨、压片,采用XRD 仪对样品进行分析,管电流为50 mA,管电压为50 kV,扫描范围为10°~40°,扫描速度为5°/min。
采用Raman 仪对样品进行分析,样品处理同FTIR 分析,扫描次数为5 次,功率为20 mW。
(4)形态结构分析。
将样品充分研磨并超声分散在无水乙醇中处理1 h,然后吸取分散液滴在覆有碳膜的铜格栅上,最后通过红外灯烘干,采用TEM 观察待测样品的形态结构。
使用导电胶将样品固定后进行喷金处理,采用SEM 观察样品的表面形貌,加速电压为5 kV。
(5)热学性能测试。
使用DSC 仪对样品的热转变行为进行分析,首先在N2环境下,以20.0℃/min 的升温速率将样品从室温升温到180℃并在此温度下保持5 min,然后自然降温到室温以消除试样的热历史,最后再以10.0℃/min 的升温速率从室温升温至180℃,记录DSC 曲线。
使用TG 分析仪分析样品的耐热行为,在N2环境下,以10.0℃/min 的升温速率并从室温升温至600℃,记录TG 曲线。
(6)电学性能测试。
将颗粒状样品在204 MPa 下压制成尺寸为Ø25 mm×0.5 mm 的圆片,按照GB/T 11007-2008,采用双电测四探针测试仪测试样品的电导率,每个样品重复测试5 次,结果取平均值。
(7)力学性能测试。
使用平板硫化机制备力学性能测试标准样条(哑铃型拉伸样条、无缺口简支梁冲击样条),温度为200℃,压力为10 MPa,喷涂脱模剂,真空排气,80℃启模。
按照GB/T 1040.2-2006,采用电子万能试验机测试样品的拉伸性能,拉伸速率为10 mm/min;按照GB/T 1843-2008,采用冲击试验机测试样品的冲击性能,冲击速度为2.9 m/s,跨距为62 mm。每个样品重复测试5 次,结果取平均值。
2 结果与讨论
2.1 MMA/DEAM-rGO 分散液的性能
(1)分散液的稳定性。
MMA/rGO,MMA/DEAM-rGO 分散液长时间自然放置均不会出现分层现象,但是当离心速度增加到5 000 r/min 时,MMA/rGO 分散液出现明显的分层现象,如图1a 所示,rGO 从分散液中沉降至离心管底部,并且自然放置rGO 也不会自动再分散;当离心速度升高到7 000 r/min 时,MMA/DEAMrGO 分散液仍呈现稳定良好、分散均匀的分散状态,如图1b 所示。以上实验结果表明,不添加改性剂的MMA/rGO 分散液的稳定性较差,添加改性剂的MMA/DEAM-rGO 分散液的稳定性良好。分散液稳定性提高的根本原因是DEAM 中氨基与rGO 表面上羟基的氢键强相互作用[12],致使改性剂DEAM牢固附着在rGO 表面上,改性剂的疏水基团代替rGO 表面上羟基基团并朝向MMA 方向,进而改善了rGO 与MMA 的相容性。

图1 高速离心后分散液的数码相机照片
(2)分散液的分散性。
图2 为分散液的PLM 照片。由图2a 可以看出,MMA/rGO 分散液出现大量rGO 团聚现象,这是由于rGO 颗粒间团聚造成的,其根本原因在于rGO 片层之间极性官能团相互作用所致,同时也与石墨烯自身的纳米特性有关;经DEAM 改性后rGO 聚集现象明显减轻,并且MMA/DEAM-rGO 分散液的分散效果较好,如图2b 所示。以上实验结果表明,添加DEAM 作为改性剂能够显著提高分散液的分散性,其根本原因是通过DEAM 中的氨基与rGO 表面上羟基的氢键强相互作用削弱了rGO 片层之间的相互作用[12],并借助DEAM 分子的亲油性提高了rGO 与MMA 的相容性,这与改性剂改善分散液的稳定性原因是一致的。

图2 分散液PLM 照片
2.2 结构与形态
(1)化学结构。
rGO,PMMA,PMG1,PMG2 和DEAM 的FTIR 谱图如图3 所示。
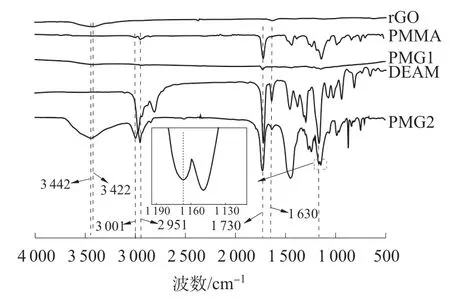
图3 rGO,PMMA,PMG1,PMG2 和DEAM 的FTIR 谱图
由图3 可知,rGO 在3 442 cm-1处出现明显的O—H 特征吸收峰,在1 630 cm-1处存在C=C 的特征吸收峰;PMMA 在1 730 cm-1处存在酯基上的C=O 特征吸收峰,在2 951,3 001 cm-1处存在甲基上C—H 的不对称伸缩振动特征吸收峰;PMG1在相同波数处的C—H 不对称伸缩振动特征吸收峰较弱,这可能是由于经丙酮萃取只残余少部分PMMA 附着在rGO 片层上所致。PMG2 在2 951,3 001 cm-1处仍出现了PMMA 的C—H 不对称伸缩振动特征吸收峰,这是由于DEAM 与rGO 间的氢键强相互作用使得PMMA 被锚定在rGO 表面上并且不受丙酮萃取的影响,DEAM 能够参与MMA的聚合反应,这从1 730 cm-1处吸收峰没有明显减弱也能得到证明。PMG2 在1 166 cm-1处仍存在DEAM 的C—N 特征吸收峰,进一步验证了DEAM与rGO 之间确实存在氢键强相互作用。另外,DEAM 对rGO 进行改性并附着在rGO 表面,然后DEAM 与MMA 发生共聚合反应,致使改性rGO 的O—H 吸收峰相对纯rGO 发生少许红移,说明rGO表面上的羟基与DEAM 中的氨基是通过氢键强相互作用将PMMA 复合到体系中的。
rGO,PMMA,PMG1 和PMG2 的XPS 谱图如图4 所示。

图4 rGO,PMMA,PMG1 和PMG2 的XPS 谱图
由图4 可知,4 条谱线均出现了两个明显的电子结合能吸收峰,分别是532 eV 的O 峰和284 eV的C 峰。rGO,PMMA,PMG1 和PMG2 的C 元素与O 元素电子结合能吸收峰强度比值(IO/IC)分别为0.27,1.14,0.91,1.18,对比可以发现,rGO 中C 元素含量相对较高,PMMA 中O 元素含量相对较高,经丙酮萃取后的PMG1,PMG2 的O 元素电子结合能吸收峰仍然较强,说明PMG1 和PMG2 中仍有少量PMMA 未被萃取完全。PMG1 中大部分PMMA被丙酮萃取除去,而PMG2 中只有少部分PMMA被丙酮萃取除去,而且PMG2 在400 eV 处还出现了较弱的N 元素吸收峰,说明DEAM 及PMMA 分子链牢固粘附在rGO 表面,并且不受丙酮萃取的影响,进一步验证了DEAM 与rGO 之间存在氢键强相互作用。
rGO,PMMA,PMG1 和PMG2 的XRD 曲线如图5 所示。
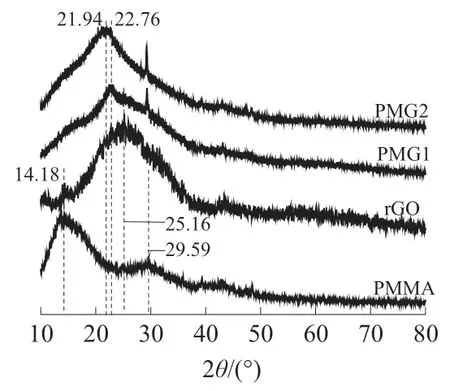
图5 rGO,PMMA,PMG1 和PMG2 的XRD 曲线
由图5 可知,rGO 在25.16°处存在较宽的衍射峰,PMMA 在14.18°和29.59°存在衍射峰,而PMG1 和PMG2 的衍射峰均向小角度方向移动,PMG1 的衍射峰在22.76°处,PMG2 的衍射峰在21.94°处。PMG1 中rGO 片层之间的PMMA 被大部分萃取除去,由于PMMA 含有大量含氧基团,经丙酮萃取后rGO 片层之间含氧基团减少,致使rGO 片层之间的范德华力增强、rGO 片层间距降低较大,最终几乎接近纯rGO 片层间距;而PMG2 中的rGO 与DEAM 的氢键强相互作用致使一部分PMMA 残留在rGO 片层之间,导致rGO 片层之间含氧基团较多,致使rGO 片层之间的范德华力增强不多,rGO 片层间距降低不大。上述实验结果表明,DEAM 改性rGO 使其表面化学结构发生改变,成功实现了MMA 在rGO 片层间的渗入以及PMMA 插层聚合物的形成,按照布拉格方程计算,PMG1 中rGO 的层间距<PMG2 中rGO 的层间距。
rGO,PMG1 和PMG2 的Raman 谱图如图6 所示。

图6 rGO,PMG1 和PMG2 的Raman 谱图
通过计算,得到图6 中rGO,PMG1 和PMG2的D 峰强度与G 峰强度的比(ID/IG)分别为0.95,1.05 和1.06,可以看出,PMG1 和PMG2 的缺陷程度均大于rGO,这些缺陷是由于MMA 渗入rGO 片层之间并发生聚合反应生成PMMA 于rGO 片层之间或包裹在rGO 片层表面大幅增加rGO 的缺陷程度造成的;PMG1 的缺陷程度小于PMG2,这是因为经丙酮萃取后PMG1 中大部分PMMA 被萃取除去,PMG2 中由于DEAM 对rGO 表面改性以及与MMA 发生共聚合反应并进入rGO 片层之间难以被萃取除去导致缺陷程度进一步增加。
(2)形态结构。
PMMA,PMMA/rGO 和PMMA/DEAM-rGO 的SEM 照片如图7 所示。

图7 PMMA,PMMA/rGO 和PMMA/DEAM-rGO 的SEM 照片
由图7a 可知,PMMA 呈现球粒形态,尺寸在1 μm 左右;图7b 中球粒结构是PMMA、灰色褶皱片层结构是rGO,rGO 在PMMA 基体中分散较为均匀,没有出现rGO 大量聚集情况,说明采用DEAM 对rGO 表面改性提高了rGO 在PMMA 基体中的分散性;图7c 中存在大量rGO 聚集以及不均匀分布现象,说明在PMMA/rGO 纳米复合材料中未改性rGO 是不均匀地分散于PMMA 基体中的。
rGO,PMG1 和PMG2 的TEM 照片如图8 所示。

图8 rGO,PMG1,PMG2 的TEM 照片
在图8a 的rGO 中,相互叠加呈不均匀分布的是rGO (黑色区域),rGO 结构发生变形扭曲,表面产生大量褶皱,其原因可能是rGO 大的比表面积发生团聚堆叠所致,也可能是rGO 界面之间存在极强的范德华力使其重复堆叠所致。在图8b 的PMG1中,仅能看到褶皱的rGO 片层结构,PMMA 近乎被丙酮萃取完全除去,说明未改性rGO 与PMMA之间的相互作用较弱。在图8c 的PMG2 中,仍能看到大范围黑色区域,其间夹杂着大量球粒形状PMMA 粘附在rGO 表面,说明rGO 表面经DEAM改性后,rGO 与PMMA 间的相互作用力增强,即使经过丙酮萃取PMMA 也没有被除去,PMMA 被牢固的锚定在rGO 片层之上。
2.3 热学性能
PMMA 及其纳米复合材料的DSC 曲线如图9所示。

图9 PMMA 及其纳米复合材料的DSC 曲线
由图9 可以看出,PMMA 的玻璃化转变温度(Tg)为102℃,PMMA/rGO 纳米复合材料的Tg为111 ℃,相对PMMA,PMMA/rGO 纳米复合材料的Tg提高了9 ℃,这主要源于rGO 的高比表面积对PMMA 大分子链段热运动的阻碍作用所致;PMMA/DEAM-rGO 纳米复合材料的Tg达到114.5℃,相对PMMA 和PMMA/rGO 纳米复合材料分别提高了12.5℃和3.5℃,通过DEAM 改性极大地提高了rGO 与PMMA 的相容性,实现了rGO在PMMA 基体中的均匀分散,加之DEAM 还可以参与MMA 的聚合反应,经DEAM 改性的rGO 在PMMA 基体中起到了物理交联点的作用,因此经DEAM 改性的rGO 对PMMA 大分子链段热运动的阻碍作用越发明显,因此PMMA/DEAM-rGO 纳米复合材料的Tg进一步得到提高。rGO 的引入提高了PMMA 的耐热性能,并且经DEAM 改性的rGO对提高PMMA 的耐热性能效果更佳。
PMMA 及其纳米复合材料的TG 曲线如图10所示。
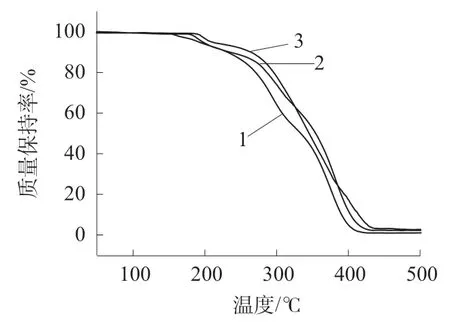
图10 PMMA 及其纳米复合材料的TG 曲线
由图10 可知,PMMA 在150.00℃开始热失重(失重5%),PMMA/rGO 纳米复合材料在179.84℃开始热失重(失重5%),相对PMMA 提升了29.84 ℃;PMMA/DEAM-rGO 纳米复合材料在187.28℃才开始热失重(失重5%),相对PMMA/rGO 纳米复合材料又提高了7.44 ℃。PMMA,PMMA/rGO 和PMMA/DEAM-rGO 纳米复合材料均存在两个热失重阶段,PMMA 的第一热失重阶段约在215 ℃左右终止,而PMMA/rGO 和PMMA/DEAM-rGO 纳米复合材料则于256℃左右终止;PMMA 第二热失重阶段始于233℃左右,而PMMA/rGO 和PMMA/DEAM-rGO 纳米复合材料则始于262℃左右,三者的热失重终止温度基本相当。当温度为435.38℃时,PMMA,PMMA/rGO 和PMMA/DEAM-rGO 纳米复合材料的烧蚀残炭率分别为1.02%,2.26%和2.71%,显然高于rGO 的添加量,这可能是因为少量CaCO3在悬浮聚合过程中被包裹在产物内部所致,也可能是因为rGO 焦化层对PMMA 包裹抑制了PMMA 的热降解。rGO 经DEAM 改性提高了其与PMMA 的相容性,更大程度地改善了PMMA 的耐热性能。
2.4 电学性能
PMMA 及其纳米复合材料的电导率见表1。

表1 PMMA 及其纳米复合材料的电导率 S/cm
由表1 可看出,PMMA 的电导率为1×10-14S/cm,属于绝缘体;PMMA/rGO 纳米复合材料的电导率达到6.78×10-4S/cm,PMMA/DEAM-rGO 纳米复合材料的电导率更是达到7.78×10-4S/cm,导电性能进一步提高。这主要源于rGO 在PMMA 基体内形成导电通道所致,PMMA/rGO 纳米复合材料完全达到防静电材料的标准,并从绝缘体转变为半导体材料。通过对比三者的电导率不难发现,添加经DEAM 改性的rGO 效果好于未改性rGO,这主要源于DEAM 的引入降低了rGO 片层间因强范德华力作用导致的团聚,对rGO 在PMMA 基体中的分散起到了很好的增容作用,大幅度地提高了rGO 在PMMA 基体中的分散均匀性。
2.5 力学性能
PMMA 及其纳米复合材料的应力-应变曲线如图11 所示,力学性能见表2。

表2 PMMA 及其纳米复合材料的力学性能
对比图11 中PMMA,PMMA/rGO 和PMMA/DEAM-rGO 纳米复合材料的应力-应变曲线初始应变区域的斜率可以看出,其拉伸弹性模量从大到小的顺序为PMMA/DEAM-rGO 纳米复合材料>PMMA/rGO 纳米复合材料>PMMA,表明在普弹性应变区间的拉伸能力从大到小的顺序为PMMA/DEAM-rGO 纳米复合材料>PMMA/rGO 纳米复合材料>PMMA。众所周知,添加未改性rGO 可以提高PMMA 的刚性,而添加改性rGO 不仅改善了其在PMMA 基体中的分散性,而且提高了其与PMMA 的相容性,实现了PMMA/DEAM-rGO 纳米复合材料刚性的进一步提高。对比三条应力-应变曲线所包围的面积以及三个试样的断裂伸长率不难发现,三个试样的韧性从大到小的顺序为PMMA/DEAM-rGO 纳米复合材料>PMMA/rGO 纳米复合材料>PMMA,表明rGO 和DEAM 改性rGO 的引入均可以提高试样的韧性,而DEAM 改性rGO改善韧性的效果更好。PMMA/rGO 和PMMA/DEAM-rGO 纳米复合材料均有屈服点(韧性破坏),并且拉伸屈服强度均高于PMMA 的拉伸断裂强度(脆性破坏),添加未改性rGO 可以提高PMMA 的韧性,实现了材料断裂方式的脆韧转变,而添加改性rGO 对促进材料脆韧转变更加显著。
由表2 可以看出,PMMA,PMMA/rGO 和PMMA/DEAM-rGO 纳米复合材料的拉伸强度依次为12.76,23.89,30.72 MPa,PMMA/rGO 纳米复合材料相对PMMA 提高了87.23%,PMMA/DEAMrGO 纳米复合材料相对PMMA 提高了140.75%。相对于添加未改性rGO 试样的拉伸强度,添加DEAM 改性rGO 试样的拉伸强度提高幅度更大。这主要源于DEAM 改性rGO 与PMMA 基体具有更好的相容性,通过DEAM 的表面改性和助分散作用,使得rGO 在PMMA 基体中能够均匀分散并起到了物理交联点的作用。PMMA 的冲击强度为3.15 kJ/m2;PMMA/rGO 纳米复合材料的冲击强度为3.71 kJ/m2,相对PMMA 提高了17.78%;PMMA/DEAM-rGO 纳米复合材料的冲击强度为3.88 kJ/m2,相对PMMA,PMMA/rGO 纳米复合材料分别提高了23.17%,4.58%。rGO 的加入大幅度提高了PMMA 的冲击性能,添加DEAM 改性的rGO 对提高PMMA 冲击性能的效果更好,这主要是由于DEAM 改性的rGO 在PMMA 基体中起到了物理交联点的作用,显著提高了PMMA/DEAMrGO 纳米复合材料中rGO 与PMMA 基体的界面相互作用力,实现了内应力在整个体系中的快速传递和均匀分散,使PMMA/DEAM-rGO 纳米复合材料的韧性和强度均得到大幅度提高。
PMMA 的断裂伸长率为2.12%,PMMA/rGO纳米复合材料断裂伸长率为2.60%,相对于PMMA提高了0.48%;PMMA/DEAM-rGO 纳米复合材料的断裂伸长率为2.93%,相对于PMMA 提高了0.81%。这与拉伸强度和冲击强度的变化规律是一致的。
3 结论
(1)DEAM 能够显著提高MMA/DEAM-rGO 分散液的分散性和稳定性,DEAM 中的氨基与rGO表面上羟基的氢键强相互作用削弱了rGO 片层之间的相互作用,提高了rGO 与MMA 的相容性。
(2)PMMA/DEAM-rGO 纳米复合材料的拉伸强度、冲击强度和断裂伸长率相对PMMA 分别提高了140.75%,23.17%和0.81%,其耐热性能同步提高,DEAM 改性rGO 在PMMA 基体中起到了物理交联点的作用,其对PMMA 大分子链段热运动的阻碍作用更明显。
(3)PMMA/DEAM-rGO 纳米复合材料的电导率为7.78×10-4S/cm,达到防静电材料标准,rGO经DEAM 改性,其在PMMA 基体中的分散性和相容性显著提高,降低了rGO 片层间因强范德华力作用导致的团聚,其在PMMA 基体内形成高效导电通道。
