氯氰菊酯乳油4种色谱分析方法比较
2022-09-24许艳秋
马 涛,许艳秋
(1.山西省检验检测中心(山西省标准计量技术研究院),山西 太原 030025;2.四川省农药检定所,四川 成都 610045)
1 前言
氯氰菊酯化学式为C22H19Cl2NO3,相对分子量:416.3,CAS号:71697-59-1,结构式如下:

氯氰菊酯原药为无味晶体,熔点41.2~47.3℃,水中溶解度极小,易溶于多数有机溶剂。稳定性:中性和弱酸性条件下稳定,碱性条件下易水解[1]。氯氰菊酯的分子结构上有3个不对称碳原子,有8个异构体[2]。是一种中等毒性杀虫剂,使用安全,在我国应用广泛,国内取得农药登记厂家众多。
目前,氯氰菊酯质量分数检测采用的是正相液相色谱内标法(方法一)或者正相液相色谱外标法(方法二)。但在日常应用中,液相色谱法绝大多数应用为反相方法,在仪器配备数量不足的情况下,需要应用正相液相色谱法时,操作人员要拆开液相色谱仪核心部件(泵)更换适配的泵杆密封圈等操作,还需要耗费大量的时间用价格较高的色谱纯异丙醇冲洗系统,工序繁琐、费时费力,操作要求和难度高于反相方法。为此,在实际工作中探索了应用相对简便、经济和环保性更好的反相液相色谱外标法(方法三)和气相色谱内标法(方法四),是较为实用的分析方法。
2 试验部分
2.1 方法一[3]正相高效液相色谱内标法
2.1.1 试剂和溶液 正己烷:色谱纯;乙酸乙酯:色谱纯;苯甲酸甲酯:分析纯(不含干扰分析的杂质);氯氰菊酯标准品:已知质量分数98.4%;5%氯氰菊酯乳油。
2.1.2 仪器 液相色谱仪:Agilent1200,具有DAD检测器和自动进样器;Agilent色谱工作站;色谱柱:150mm×4.6mm(id)不锈钢柱,内装ZORBAX RX-SIL、5μm填充物;超声波清洗机;过滤器:滤膜孔径0.45μm。
2.1.3 液相色谱操作条件 流动相:ψ(正己烷:乙酸乙酯)=99:1(V/V),用移液管移取10mL乙酸乙酯置于990mL正己烷中,摇匀,经0.45μm滤膜过滤,超声15min;内标溶液:称取0.38g苯甲酸甲酯于100mL容量瓶中,用流动相溶解并定容,摇匀;流量:1.0mL/min;柱温:室温,30℃;检测波长:278nm;进样体积:10μL;苯甲酸甲酯的保留时间约4.5min,低效顺式体的保留时间约 6.9min,高效顺式体的保留时间约8.0min,低效反式体的保留时间约8.9min,高效反式体的保留时间约10.0min。
典型的内标物和氯氰菊酯标样4个有效成分及对应的氯氰菊酯乳油的正相液相色谱图(图1)。
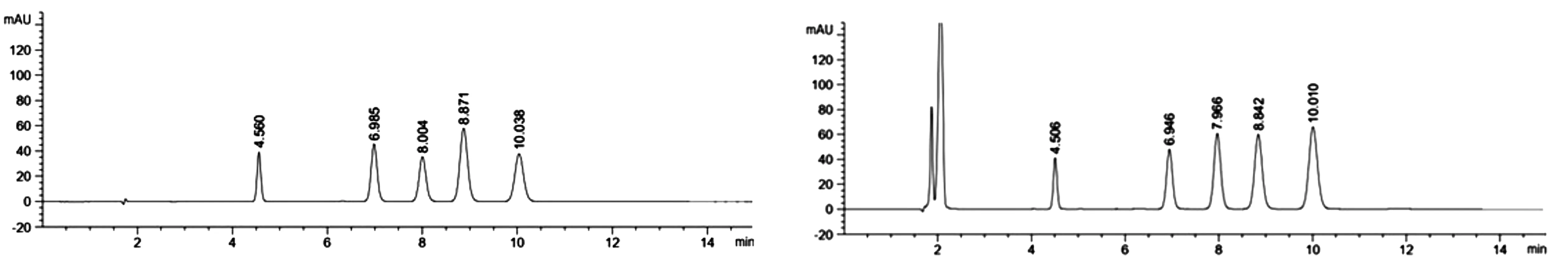
图1 内标物和氯氰菊酯标样(左)及氯氰菊酯样品(右)正相液相色谱图
2.1.4 测定步骤
2.1.4.1 标准溶液的配制 称取氯氰菊酯标样0.05g(精确至0.000 2g),于15mL具塞玻璃瓶中,用移液管加入10mL内标溶液,摇匀,备用。
2.1.4.2 试样溶液的配制 称取含氯氰菊酯的试样1.0 g(精确至0.000 2g),于15mL具塞玻璃瓶中,用配制标样溶液的同一支移液管加入10mL内标溶液,摇匀,过0.45μm滤膜,备用。
2.1.4.3 测定 在上述操作条件下,待仪器基线稳定后,连续注入数针标样溶液,直至相邻2针氯氰菊酯峰总面积与内标物峰面积之比的相对变化≤1.0%后,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行分析测定。
2.1.4.4 计算 采用内标法计算。将测得的2针试样溶液以及试样前后2针标样中的氯氰菊酯总峰面积与内标物的峰面积之比分别进行平均。试样中氯氰菊酯的质量分数ω(%)按下式计算:
(1)
式中:
r1—标样溶液中氯氰菊酯峰总面积与内标物的峰面积之比的平均值;
r2—试样溶液中氯氰菊酯峰总面积与内标物的峰面积之比的平均值;
m1—标样氯氰菊酯的质量,g;
m2—试样的质量,g;
p—标样氯氰菊酯的质量分数,%。
2.2 方法二[3]正相高效液相色谱外标法
2.2.1 试剂和溶液 正己烷:色谱纯;无水乙醚:色谱纯;氯氰菊酯标准品:同2.1.1;5%氯氰菊酯乳油:同2.1.1。
2.2.2 仪器 同2.1.2。
2.2.3 液相色谱操作条件 流动相:ψ(正己烷:无水乙醚)=98:2(V/V),用移液管移取20mL无水乙醚置于980mL正己烷中,摇匀,经0.45μm滤膜过滤,超声15min;流量:1.0mL/min;柱温:室温,30℃;检测波长:230nm;进样体积:10μL;低效顺式体的保留时间约7.1min,高效顺式体的保留时间约8.2min,低效反式体的保留时间约9.1min,高效反式体的保留时间约10.3min。
典型的氯氰菊酯4个有效成分标样及对应的氯氰菊酯乳油的正相液相色谱图(图2)。

图2 氯氰菊酯标样(左)和氯氰菊酯样品(右)正相液相色谱图
2.2.4 测定步骤
2.2.4.1 标准溶液的配制 称取氯氰菊酯标准品0.03g(精确至0.000 2g)于25.0mL容量瓶中,用正己烷定容,于超声波振荡器中震荡5min,使其全部溶解,冷却至室温过0.45μm滤膜,备用。
2.2.4.2 试样溶液的配制 称取含氯氰菊酯的试样0.6g(精确至0.000 2g)于25.0mL容量瓶中,用正己烷定容,于超声波振荡器中震荡5min,使其全部溶解,冷却至室温过0.45μm滤膜,备用。
2.2.4.3 测定 在上述操作条件下,待仪器基线稳定后,连续注入数针标样溶液,直至相邻2针标样溶液的响应值相对变化≤1.5%后,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行测定。
2.2.4.4 计算 采用外标法计算。将测得的2针试样溶液以及试样前后2针标样中的氯氰菊酯峰面积分别进行平均。试样中氯氰菊酯的质量分数ω(%)按下式计算:
(2)
式中:
A1—标样溶液中氯氰菊酯峰面积的平均值;
A2—试样溶液中氯氰菊酯峰面积的平均值;
m1—标样氯氰菊酯的质量,g;
m2—试样的质量,g;
p—标样氯氰菊酯的质量分数,%。
2.3 方法三 反相高效液相色谱外标法
2.3.1 试剂和溶液 甲醇:色谱纯;超纯水:电导率18.2MΩ.cm(25℃);其余同2.1.1。
2.3.2 仪器 液相色谱仪:Agilent1100,具有DAD检测器和自动进样器;其余同2.1.2。
2.3.3 液相色谱操作条件 流动相:ψ(甲醇:水)=88:12(V/V);流量:1.0mL/min;柱温:30℃;检测波长:230nm;进样体积:10μL;氯氰菊酯的保留时间约8min-10min。
典型的氯氰菊酯标样及氯氰菊酯乳油的反相液相色谱图(图3)。

图3 氯氰菊酯标样(左)和氯氰菊酯样品(右)反相液相色谱图
2.3.4 测定步骤
2.3.4.1 标准溶液的配制 称取氯氰菊酯标准品0.03g(精确至0.000 2g)于50.0mL容量瓶中,用甲醇定容,于超声波振荡器中震荡5min,使其全部溶解,冷却至室温过0.45μm滤膜,备用。
2.3.4.2 试样溶液的配制 称取氯氰菊酯乳油试样0.6g(精确至0.000 2g)于50.0mL容量瓶中,用甲醇定容,于超声波振荡器中震荡5min,使其全部溶解,冷却至室温过0.45μm滤膜,备用。
2.3.4.3 测定 同2.2.4.3。
2.3.4.4 计算 同2.2.4.4。
2.4 方法四 气相色谱内标法
2.4.1 试剂和溶液 丙酮:色谱纯;邻苯二甲酸二环己酯:分析纯(不含干扰分析的杂质);其余同2.1.1。
2.4.2 仪器 气相色谱仪:Agilent7890,具有FID检测器和自动进样器;Agilent色谱工作站;色谱柱:HP-5 30mm×0.32mm(id)×0.25μm毛细柱;超声波清洗机:天津奥特赛恩斯仪器有限公司AS20500BDT;氢气发生器;空气发生器;过滤器:同2.1.2。
2.4.3 气相色谱操作条件 进样口温度:270℃;进样模式:分流;分流比:50:1;载气:高纯氮气;柱流量:1.5 mL/min;柱流量控制模式:恒定流量模式;柱温:260℃,保持15min;检测器温度:270℃;氢气流量:30.0mL/min;空气流量:300.0mL/min;尾吹气流量:30.0mL/min;进样量:1.0μL;保留时间:氯氰菊酯约8.0min,内标物约4.4min。内标溶液:称取0.25g邻苯二甲酸二环己酯于25mL容量瓶中,用丙酮定容,摇匀,备用。
典型的内标物和氯氰菊酯标样及氯氰菊酯样品的气相色谱图(图4)。
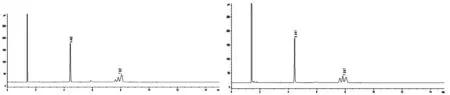
图4 内标物和氯氰菊酯标样(左)和氯氰菊酯样品(右)气相色谱图
2.4.4 测定步骤
2.4.4.1 标准溶液的配制 称取氯氰菊酯标样0.065g(精确至0.000 2g),于10.0mL容量瓶中,用移液管准确加入5.0mL内标溶液,再用丙酮定容,超声10min,静置,摇匀,加入进样小瓶时,稀释3倍。
2.4.4.2 试样溶液的配制 称取氯氰菊酯试样1.3g(精确至0.000 2g),于10.0mL容量瓶中,用2.4.4.1中同一支移液管加入5.0mL内标溶液,再用丙酮定容,超声10min,静置,摇匀,过0.45μm滤膜,加入进样小瓶时,稀释3倍。
2.4.4.3 测定 同2.1.4.3
2.4.4.4 计算 同2.1.4.4
3 结果与分析
3.1 色谱条件的选择 氯氰菊酯分析检测不需要拆分异构体,因而可以采用不能完全分离异构体的反相液相色谱法和气相色谱法。
3.1.1 液相色谱法方法条件的选择 从氯氰菊酯的紫外波长扫描图(图5)可以看到,氯氰菊酯有两个吸收峰,最大吸收波长在205nm附近,检测波长可选择范围较广。
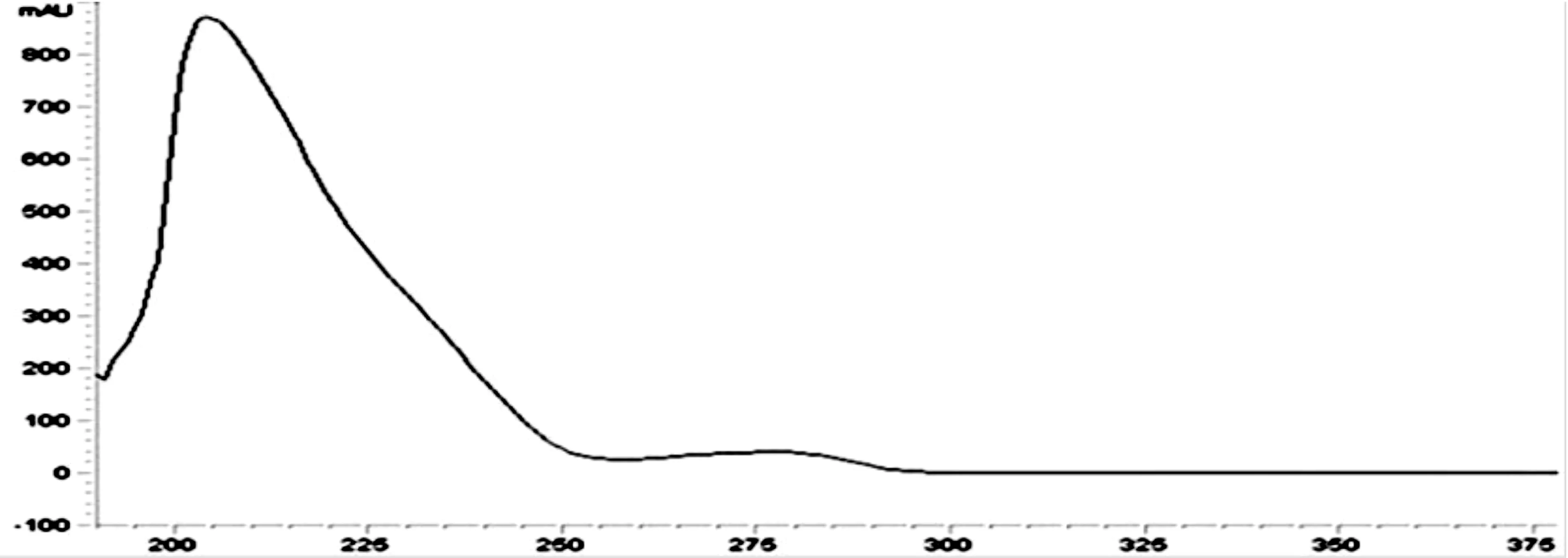
图5 氯氰菊酯紫外吸收光谱图
由于氯氰菊酯在常规溶剂中均能很好地溶解,且相对稳定,流动相选择常规溶剂即可,根据相似相溶原理,调整流动相的极性,得到各组份合适的出峰时间。
3.1.2 气相色谱方法条件的选择 气相色谱方法条件,主要的有色谱柱、进样口和柱箱及检测器温度、内标物的选择等,3个温度值都要高于待测组分的沸点,选择HP-5色谱柱及邻苯二甲酸二环己酯作为内标物,可获得满意的分析结果。
3.2 精密度测定 采用上述4种方法测定同一5%氯氰菊酯乳油产品,每种方法平行测定6次,测定各方法的精密度。测定结果(表1),方法一、方法二、方法三、方法四4种方法的平均值分别为4.99%、5.01%、5.00%、5.04%,标准偏差分别为0.080、0.085、0.090、0.083,变异系数分别为1.61%、1.70%、1.80%、1.65%。
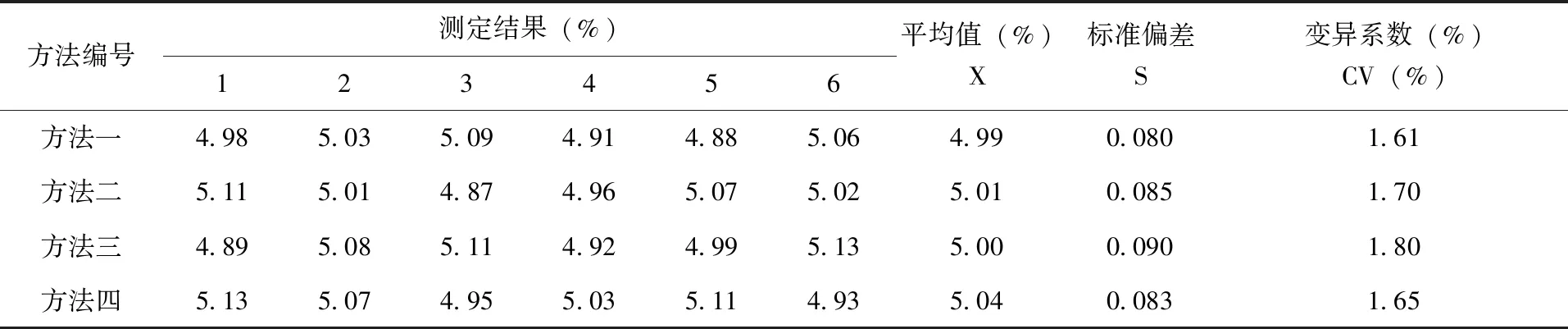
表1 4种方法分析5%氯氰菊酯乳油精密度数据
3.3 准确度测定 准确度测定采用标准加入法,分别称取已知含量的5%氯氰菊酯乳油20份,各加入一定量的氯氰菊酯标准溶液,每一种方法测定5份加标后的样品,测定其添加量的回收率。测定结果(表2),可见方法一到四4个方法回收率范围分别在99.04%~101.61%、98.81%~101.35%、98.02%~100.84%、99.55%~101.20%之间。

表2 4种方法分析5%氯氰菊酯乳油准确度数据
3.4 线性关系测定 2种内标法分别用各自配好的标准溶液,在各自色谱条件下分析。用氯氰菊酯与内标物质量比值(0.439、0.877、1.316、1.756、2.195五个水平)做横坐标,氯氰菊酯与内标物峰面积比值做纵坐标。得到正相液相色谱内标法线性回归方程y=4.847 9x+0.069 3,R2=0.999 4,相关系数r=0.999 7;气相色谱内标法线性回归方程y=0.7302x-0.008 9,R2=0.999 5,相关系数r=0.999 7(表3、图6)。

表3 方法一、四线性关系数据表

图6 方法一(左)和方法四(右)线性关系图
2种液相色谱外标法,分别用各自配好的标准溶液,在对应色谱条件下分析,进样量分别为1、2、5、10、15、20μL,以氯氰菊酯浓度值做横坐标,氯氰菊酯峰面积为纵坐标,制作线性关系曲线。得到正相液相色谱外标法线性回归方程y=5 083x-7.553 2,R2=0.999 2,相关系数r=0.999 6;反相液相色谱外标法线性回归方程y=53 514x+60.297,R2=0.999 9,相关系数r=0.999 9(表4、图7)。

表4 方法二、三线性关系数据表

图7 方法二(左)和方法三(右)线性关系图
3.5 4种分析方法结果比较 按照NY/T 2887-2016《农药产品质量分析方法确认指南》要求[4],对于5%氯氰菊酯乳油产品,分析方法精密度要求变异系数<2.11%(计算公式:2(1-0.5logC)×0.67,此处C=0.05)、线性系数>0.99、添加回收率要求在97%~103%之间才合乎要求。从表1、2、3看出,4种方法精密度、准确度、线性关系均符合要求,分析所用时间合适,均能满足日常分析要求。
表5就4种分析方法经济性、环保性、便捷性和操作难度四个方面做比较。可看出,气相色谱法因流动相为高纯氮气,所以经济性和环保性最好,操作难度小,便捷性较好;反相液相色谱法最便捷,成本也低于正相液相色谱法;正相液相色谱法既不经济也不环保,更不便捷,操作难度大,非熟练技术人员比较难获得好的数据结果。

表5 4种方法各项性能比较
4 结论
综上所述,在实际应用中,推荐使用气相色谱内标法或者反相液相色谱外标法来完成氯氰菊酯乳油含量的检测工作。
