单细胞与空间转录组分析研究进展
2022-09-21蔡林峰张倩楠杨启正梁珊珊李铭殷杨朝勇
许 醒,蔡林峰,张倩楠,杨启正,梁珊珊,李铭殷,李 翀,杨朝勇,2*
(1.厦门大学 化学化工学院,福建 厦门 361005;2.上海交通大学 分子医学研究院,上海 200217)
17世纪中叶,荷兰显微镜学家列文虎克与英国科学家胡克发现了细胞,揭开了生命体基本单位—细胞的面纱。由于细胞增殖、分化和代谢等过程受到细胞内在生命活动以及周围微环境的影响,不同细胞在细胞大小、化学组成、生物活性、生理响应对象以及响应时间等方面存在异质性。为了解码各种类型细胞的身份、特征和功能状态,单细胞分析应运而生,其通过对单个细胞的形态学、遗传学分析,从而了解不同组织、器官及生命体的特征,探究不同生命活动的发生过程。单细胞测序是单细胞分析中的重要组成部分,其以细胞内的RNA、DNA等核酸分子作为研究对象,利用测序技术进行结构与序列解析,为细胞异质性研究提供可靠、准确的研究手段及观测视野,为揭示生物发育规律、疾病发生发展机制等提供了重要的技术手段[1-2]。2019年,“单细胞多组学技术”被Nature Methods评为年度技术方法;2020年,“空间转录组技术”被Nature Methods评为年度技术方法;2022年,空间多组学被Nature评为值得关注的七大技术之一。至此,单细胞测序分析朝着多维度、空间化方向发展。本文将围绕单细胞和空间转录组分析,系统讨论近几年单细胞转录组测序、单细胞多组学测序及空间转录组分析的研究进展,并总结及展望其未来的发展方向。
1 单细胞转录组测序分析
单细胞转录组测序(scRNA-seq)通过对单个细胞内的RNA(~10 pg)进行提取及文库制备,利用高通量测序技术读取文库序列获得全转录本信息,以辨别细胞类型、状态与功能,阐明细胞异质性及进化轨迹等。单细胞转录组测序靶标检测通量高,理论上可实现对单细胞全转录组的同时测量,提高了对单细胞异质性的辨别能力;同时,转录组不仅继承了基因组的遗传信息,还反映了细胞基因选择性表达的详细情况,细胞分型准确性高。目前,单细胞转录组测序已在肿瘤生物学、发育生物学、神经生物学、微生物学等多个领域的研究中得到广泛应用[3-6]。单细胞转录组测序主要流程包括单细胞的分离、单个细胞核酸的提取、微量核酸的扩增与建库等。根据文库类型差异分为基于全长转录组的文库构建法和基于标签转录组的文库构建法。
1.1 基于全长转录组的文库构建法
该文库构建方法针对全长转录组,对基因、单核苷酸多态性、可变剪切体具有高检测能力,并易于定位来源细胞。2009年,汤富酬课题组首次报道了单细胞转录组测序方法[7]。该方法利用末端转移酶将poly(A)短链添加至逆转录合成的第一链cDNA上,再通过杂交锚定有聚合酶链式反应(PCR)引物的poly(T)短链完成第二链合成,进而进行PCR扩增,首次实现了单细胞维度的基因表达异质性研究。Sandberg课题组于2014年发展的Smart-seq2技术是目前应用最广的单细胞全长转录组测序技术之一。其利用M-MLV逆转录酶同时具有模板转化与末端转移酶活性的特点,在cDNA合成至3′端时在其尾部添加一段特殊序列并以此作为引物序列实现PCR扩增[8]。该课题组于2020年提出了Smart-seq3技术,在cDNA合成过程中引入独特分子标记(UMI),通过校正扩增偏差实现了对RNA的准确计数,提高了基因检出的灵敏度与准确度[9]。
然而,以上基于离心管的建库方式存在细胞挑取困难,试剂消耗大,核酸反应效率低等问题。与之相比,微流控技术可提供pL至nL级反应体系,与单细胞尺寸相匹配,在提高模板反应浓度的同时减少了试剂消耗量,具有高转录本检出能力及低成本的优势[10-11]。黄岩谊课题组开发了一种集成式微流控芯片用于全长scRNA-seq分析,提高了基因表达测量的准确性和灵敏度[12]。商业化平台Fluidigm C1系统使用了类似的设计,兼容Smart-seq2等文库构建方式[13]。Sarma等[14]设计了一种通过浓度梯度驱动引起流体扩散进行试剂去除和输送的微流控平台,可在同一个腔室中完成Smart-seq2全步骤。然而,以上平台使用泵/阀结构控制流体运动,需要复杂的芯片结构设计与制作工艺。为了简化微流控芯片的设计、制作与操控,增加平台的普适性,本课题组基于具有离散液滴操控功能的数字微流控芯片结合Smart-seq2技术,构筑了自动化单细胞转录组测序平台(Digital-RNA-seq,图1A),实现了快速、高效、灵敏及准确的单细胞RNA检测[15]。Digital-RNA-seq通过在芯片上构筑亲-疏水特征结构,实现了单细胞的快速、高效和无损分离;采用纳升级反应体系及疏水界面增加模板的反应浓度,减少核酸吸附,实现了高灵敏的基因检测;借助均匀的液滴生成和油相隔离的反应空间减少加样偏差及外源污染,提高了测量的准确性。相比传统离心管方法,该平台减少了近百倍的试剂消耗量,实现了低成本的单细胞转录组分析。在此基础上,本课题组进一步将文库纯化、文库构建等部分集成,实现了“细胞进,文库出”的一体化操作(Cilo-seq),将文库检出率提高了1.4倍[16]。

图1 基于数字微流控的单细胞转录组测序(digital-RNA-seq)[15](A);基于配对微流控芯片的高通量单细胞测序(paired-seq)[25](B)Fig.1 Single-cell transcriptome sequencing based on digital microfluidics(digital-RNA-seq)[15](A);high-throughput single-cell sequencing based on paired microfluidic chip(paired-seq)[25](B)
1.2 基于标签转录组的文库构建法
该文库构建方法主要通过在转录本一端添加标签标记细胞来源,从而允许高通量单细胞转录组平行分析。Yanai课题组于2016年推出CEL-Seq2技术,利用随机寡核苷酸链在逆转录步骤为每个单细胞的转录本标记上独一无二的细胞编码与分子编码,由此可在后续扩增过程中将上千个细胞的核酸在同一个反应池中进行反应,大大降低了人力、试剂与测序成本,使单个测序文库包含大量单细胞的转录组信息[17]。为了进一步提高单细胞转录组分析通量,Macosko课题组和Weitz课题组分别发展了Drop-Seq与Indrops技术,基于携带有细胞编码与分子编码的编码微球,利用液滴微流控芯片在短时间内生成大量油包水液滴,同时将单个细胞与单个微球包裹于同一微液滴中,以此实现高通量单细胞的快速分离及对应转录组信息的标记[18-19]。Shalek课题组和郭国骥课题组将该技术拓展至微阵列体系中,分别开发了Seq-Well和Microwell-seq平台,在微孔中实现了单个细胞与微球的配对、mRNA捕获、编码及逆转录,通过测序技术,获得了成千上万个单细胞的转录组表达信息[20-21]。此类方法极大提高了单细胞的分析通量,进一步降低了试剂用量和分析成本,推动了单细胞转录组测序的产业化进程,如10×Genomics公司的ChromiumTM液滴微流控平台、BD公司推出的RhapsodyTMSingle-Cell Analysis System等。然而,大多数方法由于单细胞和编码微球的包裹基于泊松分布,许多细胞无法与微球成功配对从而造成信息丢失,导致细胞利用率偏低。
为了克服泊松分布,樊荣课题组开发了一种集成的介电电泳(DEP)-捕获-纳微孔转移方法(dTNT),通过DEP辅助捕获细胞于微阵列中(捕获效率达91.84%),然后将整个装置翻转并改变DEP的方向,使捕获于DEP阵列中的单细胞落入与其对应的大孔径微阵列中(细胞转移效率为82%),以实现单细胞与单微球在微孔中的高效配对[22]。基于泵阀控制结构的微流控芯片提供了另一种提高细胞利用率的方法,可实现稀有样本包括循环肿瘤细胞、肠类器官的单细胞分析[23-24]。本课题组开发了Paired-seq技术(图1B),通过发展基于差异流阻原理的单细胞/单微珠操控微流控芯片,实现了单细胞/单微球的高效快速捕获,成功突破了因泊松分布导致的细胞利用率低的限制,在稀有细胞分析方面具有明显优势(细胞利用率>95%);同时Paired-seq利用泵阀操控结构移除游离RNA以降低背景干扰,并通过皮升级反应腔体增加单细胞mRNA的捕获能力,大大提高了转录组的基因检出效率[25]。Paired-seq的分析通量在千级水平,为了提高样品分析通量,本课题组进一步发展了一种协同尺寸排阻和局部准静态动力学的双层微孔芯片平台(Well-Paired-seq),用于超高通量的scRNA-seq分析,一个芯片可以分析超过10万个细胞。通过尺寸排阻原理允许在细胞捕获微孔和微球捕获微孔分别捕获一个细胞和一个微球,并通过准静态流体力学原理确保被捕获的细胞不被冲洗出去,从而实现细胞的累积捕获和有效的缓冲交换[26]。Well-Paired-seq保证了高密度的单细胞/微球配对,实现了优异的细胞捕获效率(~91%)、细胞/微球配对效率(~82%)和细胞游离RNA去除效率。Well-Paired-seq的超高通量分析能力、高效细胞捕获效率和高游离RNA去除效率等优势在细胞图谱绘制、细胞异质性分析、稀有细胞亚群分析中具有重要的应用价值。
细胞原位编码技术是另一类基于标签转录组的文库构建技术。该方法无需利用微液滴及微阵列对细胞进行分隔,而是将细胞本身作为单独的反应器。同时,该方法无需使用编码微球,通过组合拆分方法即可对大量单细胞进行细胞及分子编码。Shendure课题组和Seelig课题组将大量细胞分配至96或384孔板中,利用含有编码序列的引物对cDNA进行标记,其后将所有细胞回收并重新分配至含有新编码序列的引物的96或384孔板中,对cDNA进行下一轮标记,然后重复组合-拆分过程[27-28]。此时,编码容量随着组合-拆分次数的增加呈指数型增长。因此,该方法的细胞分析通量极大,但由于需要不断地进行拆分混合操作,文库构建存在繁琐性与复杂性。近期,Bock课题组将细胞原位编码技术与液滴微流控技术相结合,开发了单细胞组合流体索引技术scifi-RNA-seq[29]。该方法通过在384孔板中将透化的细胞(核)随机等分,使用孔板中的预标第一轮条形码实现cDNA的合成与标记。然后,将含有预索引cDNA的细胞(核)随机混合,使用具有高度过载的标准微流控液滴发生器进行封装,使大多数液滴包含多个细胞(核)。在这些过载的液滴中,转录本用液滴内第二轮条形码进行标记。因此,在同一分裂池中的所有细胞共享第一轮条形码,在同一液滴中的所有细胞共享第二轮条形码,两种条形码的组合可唯一编码来自同一单个细胞的转录本。与传统多轮组合拆分技术相比,scifi-RNA-seq更为简便高效;与单一液滴微流控方法相比,scifi-RNA-seq允许每个液滴内包裹多个细胞,大大提高了分析细胞通量,避免了传统液滴方法因包裹多个细胞导致的错误。
单细胞转录组测序主要朝着3个方向发展:(1)更高基因检出率;(2)更高通量;(3)更高细胞利用率。然而,目前仍面临以下两大问题:基于全长转录组的文库构建法相对于基于标签转录组的文库构建法基因检测能力更高,但是牺牲了高通量分析的能力,高基因检出率与高分析通量难以兼得;超高通量与高细胞利用率(尤其是针对稀有细胞)的分析仍难以平衡,灵活性强、样品适用范围广的操纵平台仍有待进一步开发。
2 单细胞多组学测序分析
单个细胞的表型往往受到不同分子(如DNA、RNA及蛋白质)的共同调控,单细胞单组学分析难以提供全面的细胞异质性信息。与之相比,单细胞多组学分析可以从多种分子层面揭示细胞特性,同时将不同组学信息直接关联,更加深入地研究单细胞状态、功能与生长过程及其分子调节机制[30]。目前,单细胞转录组及其与其它组学的联合分析在技术开发与整合分析方面已取得了初步进展。
2.1 单细胞转录组和基因组联合分析
基因组是决定细胞内遗传变异分子机制的源代码,而转录组决定了细胞的特定功能。基因组和转录组的联合分析通过探究DNA拷贝数、DNA编码区与非编码区如何决定转录组表达水平,从而建立基因组和转录组的相关性,并阐述选择性表达分子作用机制,如RNA编辑、调控变异和等位基因特异性表达[31]。另外,对不同细胞的基因组和转录组进行联合分析,可进一步区分遗传亚克隆,同时进行谱系示踪。单细胞转录组和基因组的联合分析步骤主要包括单细胞分离、细胞裂解、DNA/RNA分离、基因组/转录组文库构建。
DR-seq和G&T-seq是单细胞DNA和RNA平行分析的开创性工作[32-33]。DR-seq在挑取并裂解单细胞后,mRNA经测序适配子(Ad-1x)进行反转录;接着以适配子(Ad-2)为引物,进行cDNA与基因组DNA的线性预扩增;之后将产物分成两个部分,分别用于MALBAC全基因组文库构建及CEL-seq转录组文库构建。DR-seq避免了DNA和RNA的物理分离,减少了核酸的损失及操作繁琐度,但易造成DNA与RNA间的相互污染。G&T-seq利用寡聚poly(T)包被的磁珠从细胞裂解液中物理分离含poly(A)尾的mRNA,接着对DNA和mRNA分别进行MDA基因组文库构建和Smart-seq2转录组文库构建。这种单细胞DNA/RNA分离方式可适用于多种下游建库方法,亦可将所获得的DNA用于甲基化组研究。然而由于该方法针对poly(A)尾mRNA,难以应用于非编码RNA的测量。汤富酬课题组发展的ScTrio-seq采用温和裂解法,在保持细胞核完整的同时裂解细胞膜以获得所有胞质RNA,通过移取上清液实现RNA/细胞核的分离,并将细胞核内DNA用于全基因组和甲基化组的同时分析[34]。然而,为了在移取上清液的过程中避免细胞核的不慎吸取,该方法保留了部分上清液,造成RNA的损失。为了提高RNA的回收量,SIDR-seq利用抗体修饰磁珠标记单个细胞,然后采用低渗裂解策略在细胞质膜上进行穿孔以释放RNA,并通过磁分离方式实现RNA/细胞核的分离,允许不同类型的下游文库构建[34]。
然而,以上方法均局限于细胞的分析通量。为了解决这一问题,Shendure课题组采用组合拆分策略实现了高通量单细胞基因组和转录组联合分析(sci-L3,图2A)[36]。细胞被固定及核小体剥离后,分散至多孔板中;在每个孔板中,对细胞基因组进行基于Tn5酶的片段化及插入反应,对转录组进行逆转录反应,通过Tn5插入引物及逆转录引物分别对细胞的基因组及转录组标记第一轮编码;将所有细胞混合,重新随机拆分至孔板中,通过连接技术添加包含有T7启动子的第二轮编码;将所有细胞重新拆分,并进行间隙扩展形成双链T7启动子,随后经过体外转录、逆转录和第二链合成,添加第三轮编码。同一个细胞内的RNA与DNA共享相同的细胞编码,通过测序数据中不同的文库结构区分RNA/DNA来源,最终实现高通量单细胞RNA/DNA的联合分析。然而,该方法的基因组覆盖率只有20%,因此集高通量、高覆盖率于一体的单细胞基因组和转录组联合分析方法仍有待开发。
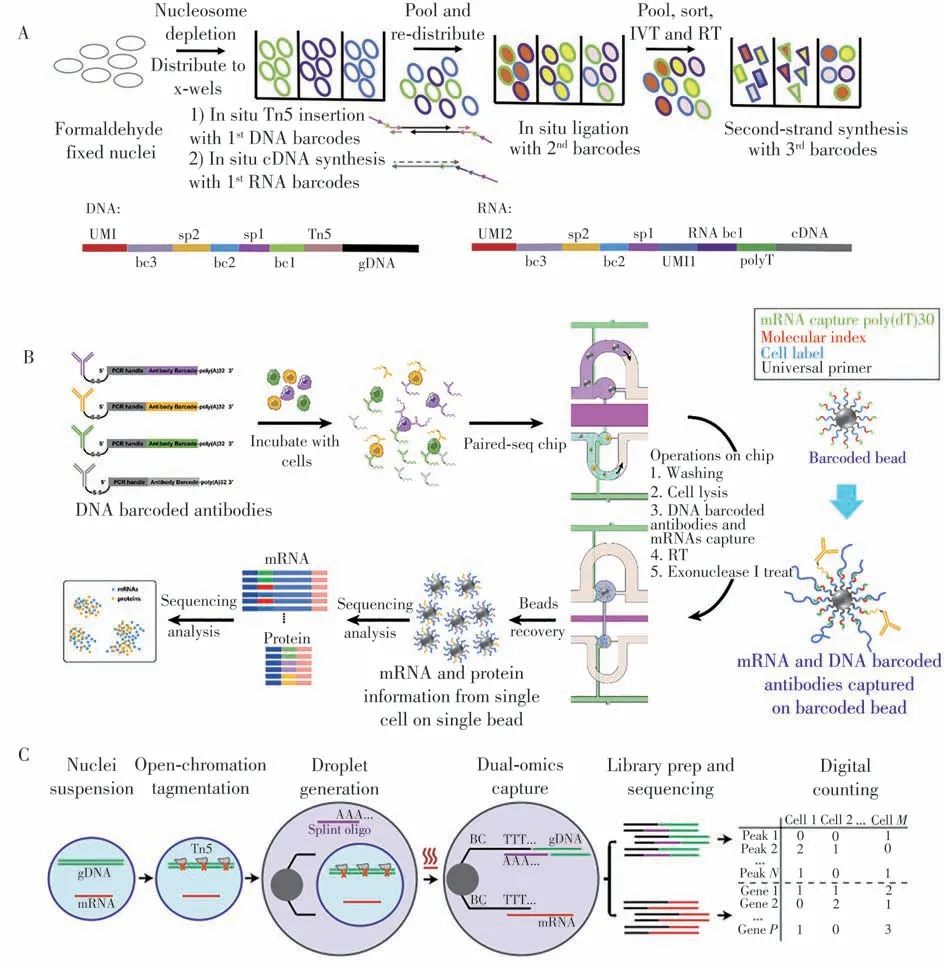
图2 高通量单细胞基因组和转录组联合测序(sci-L3)[36](A);高通量单细胞转录组和蛋白质联合测序(multi-Paired-seq)[43](B);高通量单细胞染色质可及性和转录组联合分析(SNARE-seq)[47](C)Fig.2 High-throughput single-cell genome and transcriptome sequencing(sci-L3)[36](A);high-throughput single-cell simultaneous transcriptome and proteins sequencing(multi-Paired-seq)[43](B);high-throughput single-cell chromatin accessibility and transcriptome sequencing(SNARE-seq)[47](C)
2.2 单细胞转录组和蛋白质组联合分析
蛋白质作为表型的直接输出者,直接反映细胞当下的生理状态和细胞功能。转录组和蛋白质组的联合测量有助于解释转录异质性如何转化为功能表型多样性,深入理解转录/翻译后修饰过程;同时有助于揭示RNA层面无法区分的表型差异,并探讨特定细胞亚型的基因表达网络和功能调控类型。
基于荧光探针杂交或实时定量荧光PCR的传统RNA/蛋白测量法由于受到荧光光谱重叠和引物种类限制,靶标检测及细胞分析通量有限[37-38]。基于微流控技术和高通量测序技术的CITE-seq和REAP-seq有效解决了这一问题[39-40]。该类方法利用含有抗体编码及poly(A)的DNA序列标记并偶联特定抗体,从而将蛋白信息转化为核酸信息;接着借助Drop-seq及10×Genomics技术平台在标记抗体孵育细胞后将单个细胞及编码微球共包裹于液滴中,细胞裂解所释放的mRNA和标记抗体DNA在碱基互补配对原则下被编码微球捕获。通过逆转录反应获得带有细胞编码、分子编码的cDNA与标记抗体DNA,对所有产物进行扩增与测序,首次实现了成千上万个单细胞转录组与蛋白质的联合分析,不仅提高了分析通量,且可用于低丰度蛋白检测。在此基础上,Abate课题组将DNA标记抗体更替为靶向特定蛋白的核酸适体,减少了抗体使用量并降低了成本[41]。Smibert课题组进一步将该类方法与T细胞/B细胞受体、细胞标签等分析结合,拓展了单细胞领域的多模态分析[42]。为了提高单细胞利用率,减少泊松分布带来的细胞损失,本课题组结合抗体编码与Paired-seq技术[25]发展了高通量单细胞转录组和蛋白质联合测序平台(multi-Paired-seq,图2B)[43]。该平台在标记抗体孵育细胞后将单个细胞通入微流控芯片,基于差异流阻原理进行单细胞/单微球的顺序捕获与配对;接着利用含poly(T)的编码微球捕获细胞裂解释放的mRNA及标记抗体DNA,通过逆转录、扩增、建库及测序构建单细胞转录组与蛋白质的表达矩阵。multi-Paired-seq证实了其在细胞利用水平、蛋白质检测准确性等方面的优越性,并成功揭示了单细胞在mRNA与对应蛋白间的表达差异性。
然而以上方法均局限于细胞膜蛋白的分析。为了实现细胞膜内蛋白的联合测量,Mulder课题组将人工合成的标记RNA与抗体偶联,令其进入固定后的细胞后于原位与磷酸化蛋白结合,并基于CELseq技术为每一个细胞的RNA及标记抗体的合成RNA进行编码、扩增与测序,最终实现了单细胞mRNA及膜内磷酸化蛋白的联合分析[44]。因此,当前的单细胞转录组和蛋白质联合分析正朝着高通量、多模态方向发展。然而,DNA/RNA标记抗体偶联效率有限,制备工艺繁琐,商品化的DNA标记抗体价格昂贵,可替代的核酸适体种类有限,亟需开发高效便捷的蛋白标记方法以提高单细胞转录组和蛋白质组分析的普适性与广泛性。
2.3 单细胞转录组和表观遗传组联合分析
细胞内表观遗传标记变化广泛,如DNA甲基化修饰、组蛋白修饰、转录因子结合、染色质可及性等,其通过调节染色质结构与构象促进/抑制转录组表达。因此,对转录组和表观基因组进行联合分析可以解析染色质结构与构象变化对细胞基因表达的可塑性,并构建细胞表观遗传调控网络,揭示基因表达调控机制;同时,二者的同时分析可帮助鉴别细胞发育和疾病发展过程中的特殊标记并实现遗传谱系示踪。
染色质可及性和转录组的联合检测为揭示单个细胞的基因调控机制提供了强有力的工具。Cao等基于组合拆分策略发展了Sci-CAR技术,通过将细胞核随机分配于多孔板中并依次进行原位逆转录与Tn5酶打断标记,为RNA文库及染色质可及性文库添加第一轮编码;接着将所有细胞核合并并重新分配于多孔板中,对其进行裂解后将产物分成两部分,在PCR过程中添加第二轮编码;由于来自同一细胞的RNA文库及染色质可及性文库拥有相同编码,因此可关联同一单细胞的转录组和染色质可及性并作联合分析[45]。为了进一步提高分析通量,Zhu等在Sci-CAR的基础上,增加了三轮基于连接反应的编码,将单细胞分析通量提高至1×107[46]。为了提高分析灵敏度并减少复杂操作,Chen等提出了SNARE-seq(图2C),通过在透化的细胞核内进行Tn5酶标记反应并引入与标签适配子互补的含有poly(A)的夹板序列作为介导,利用液滴微流控技术将单个细胞核与编码微球包裹于同一液滴中,使编码微球可同时捕获夹板序列和mRNA,并利用测序技术进行染色质可及性与转录组信息读取[47]。相比Sci-CAR,SNARE-seq多检测到了4~5倍的可及DNA。Greenleaf课题组使用Cy5标记DNA偶联的绿色荧光蛋白(GFP)抗体对HEK293细胞表达的核定位GFP进行染色,然后基于10×Genomics液滴微流控平台生成单细胞染色质可及性文库和转录组文库,通过一次测序即实现了对内源性核蛋白丰度、染色质可及性和转录组的定量分析[48]。然而以上方法均基于细胞核进行,牺牲了细胞质转录本;此外均将产物分成两部分以进行染色质可及性文库与转录组文库的独立制备,导致了一半信息的丢失。因此,基于全细胞水平的染色质可及性和转录组文库集中构建方法仍有待开发。
单细胞DNA甲基化组和转录组的联合分析可鉴别突变产生的遗传谱系,全面揭示细胞间的异质性,同时探讨DNA甲基化修饰对基因表达的调控规律。scM&T-seq通过流式细胞仪分选单个细胞,然后利用oligo-dT磁珠对细胞内的DNA及RNA进行物理分离,并对其分别进行甲基化文库(scRRBS)及转录组文库(Smart-seq2)构建[49]。同年报道的scMT-seq利用相似的方法对单个细胞中的DNA及RNA进行分离,并分别进行全基因组水平的甲基化文库(scWGBS)和转录组文库(Smart-seq2)构建[50]。然而,以上两种方法易在DNA/RNA的物理分离过程中造成核酸损失。为了解决这一问题,Luo等发展了单管操作snmCT-seq技术,将单个细胞分选至384孔板,在逆转录和cDNA扩增过程中用5mC-dCTP代替dCTP,因此cDNA链上全部的C碱基均为甲基化的;随后进行亚硫酸氢盐转化,发生C>T转变。而gDNA中少量胞嘧啶是甲基化的,其他胞嘧啶都是未甲基化的,因此可通过测序进行RNA及gDNA来源序列区分,从而实现单细胞转录组和甲基化组的联合分析[51]。然而该方法未事先对GC区域进行富集,在测量甲基化位点时需加大测序深度在全基因组范围内搜寻,成本昂贵。
为了进一步增加单细胞的多模态分析,构建多维表观遗传调控图谱,scNMT-seq和scChaRM-seq在DNA/RNA分离后,引入甲基转移酶标记可及的DNA,仅使开放染色质上GCH位点未甲基化的胞嘧啶全部甲基化,其他位置的甲基化状态保持不变,通过比对GpC和CpG位点同时辨别单细胞染色质可及性区域和DNA甲基化修饰[52-53]。RNA部分用于构建scRNA-seq文库,从而实现单细胞甲基化、染色质可及性及转录组三组学的联合分析。然而,目前涉及甲基化的单细胞多组学分析由于操作步骤较为繁琐,难以利用液滴微流控或组合拆分策略提高分析通量,因此高通量、高灵敏的多维表观遗传组与转录组联合分析方法开发是该领域未来的发展方向。
3 空间转录组分析
在单细胞转录组测序中,单细胞的获取依赖于组织解离,因此丢失了细胞的空间信息。空间转录组技术是结合成像、生物标记、测序及生物信息学等工具对组织切片的基因表达进行空间定位的一项技术,可揭示各细胞类型在组织中的空间分布、各细胞群体间的相互作用以及绘制不同组织区域的基因表达图谱,对于理解疾病和癌症的发生机制具有深远的应用价值[54-55]。当前的空间转录组技术主要分为激光显微切割、荧光原位杂交、荧光原位测序以及原位捕获技术四大类。
3.1 激光显微切割法
结合二代测序技术的激光显微切割法是空间转录组研究的有力工具。我国中科院景乃禾、韩敬东和彭广敦课题组发展了Geo-seq,利用激光显微切割直接分离组织中特定位点的细胞进行转录组测序,以充分获得组织中各位点的全转录组信息[56]。为了进一步实现单细胞分辨率的测量,Nichterwitz等[57]开发的LCM-seq将激光显微镜切割与Smart-seq2结合,实现了单细胞水平的空间转录组分析。结合二代测序技术的激光显微切割法在稀有组织或部分降解组织样本分析中具有优势,然而面临细胞形态破坏、空间分辨率有限、细胞逐个切割分离耗时长等问题。
3.2 荧光原位杂交法
荧光原位杂交法利用标记的荧光探针确定组织或细胞中的RNA/DNA丰度,并逐渐发展至单分子分辨水平。传统的单分子荧光杂交技术(smFISH)受到荧光光谱重叠限制,只能检测3~4种目标RNA。组合标记技术通过多种荧光标记的组合可有效提高荧光原位杂交通量。蔡龙课题组发展的seqFISH技术,在每轮杂交中利用24个标记有单一颜色的FISH探针与每个转录本杂交;成像后将探针剥离,在下一轮的杂交中使用拥有相同序列但标记不同荧光团的探针与相同的靶标杂交[58]。通过连续几轮杂交、成像和探针剥离,参与每一轮杂交的不同荧光排序形成了每个转录本的独特条形码。利用F种荧光团数目,经过N轮杂交后,seqFISH可对F×N种转录本进行条形码编码,提高了靶标检测通量。然而该方法存在因荧光信号的非特异性杂交引入的潜在错误率,这种错误随顺序杂交而累积,因此对基因表达鉴定的准确性提出了重大挑战。为了降低潜在错误率,庄小威课题组发展了MERFISH技术,应用于RNA、染色质DNA三维结构等靶标测量[59-61]。在MERFISH中,每种RNA被设计与192个编码探针杂交,该编码探针由一个靶向序列与两个读出荧光探针组成。在每一轮杂交中,通过系列的荧光探针靶向靶标RNA并读取荧光信号,产生的信号在相应的一轮中记为“1”,无荧光信号的一轮记为“0”;经过N轮杂交后,每个位点可生成由N个二进制编码所组成的条形码,通过与所设计的靶标条形码比对,获得该位点的具体靶标RNA信息。同时,MERFISH设计了一种纠错编码策略,通过设定二进制位编码的最小汉明距离以选择相差更明显的编码,在遇到1个荧光标记识别错误时可通过人为修改回归正确的编码标记,提高了转录本检测的准确性。
为了进一步提高RNA的检测通量,蔡龙课题组发展了升级版seqFISH技术(intron seqFISH)[62-63]。在该技术中,所设计的探针由一个靶向靶标基因内含子的特异性序列和4个延伸序列组成,每一段延伸序列可与对应的读出探针杂交成像。在每一轮成像中采用3组标记有不同荧光团的探针,通过对4个序列杂交的叠加图像进行数据处理,可以在单轮中获得12个伪彩色的图像。通过5轮杂交(另一轮进行误差修正),intron seqFISH可解码10 421种转录本。尽管具有高靶标检测通量,但该方法需对样品进行数十轮的杂交反应,步骤复杂、成本高。为了减少杂交次数,李景虹课题组发展了基于调控核酸杂交热力学的DNA序列编码的荧光标记技术(SeqEA,图3A)[64]。SeqEA利用含有识别单元(R)和编码单元(B1、B2....)的编码锁式探针,在R识别靶标RNA后引发滚环扩增(RCA)反应,产生一个具有几百个与锁式探针序列互补DNA的单链产物,并通过与荧光标记的检测探针P1和P2杂交实现成像。通过热力学计算指导的编码模块B1序列能够精确调控B1-P1杂交链的杂交效率,从而调控结合到RCA产物的P1数量,并利用所产生的荧光信号差异对不同转录本进行荧光编码。该方法利用DNA序列作为高密度的信息存储载体,具有可编程、模块化及分子水平编码稳定等特点,减少了荧光探针使用数目及杂交次数,可实现高通量、易操作的单分子、单核苷酸分辨率的转录本空间分析。综上所述,现有的荧光原位杂交法在空间分辨率及靶标检测通量上具有优势,但其缺点在于需要合成大量荧光探针,昂贵耗时,且仅能对已知的靶标转录本进行测量。
3.3 荧光原位测序法
荧光原位测序法利用单碱基特异性荧光杂交和DNA连接反应对组织中已知或未知转录本的序列进行原位测定。柯荣秦等发展的原位测序(ISS)技术将细胞固定透化后,在原位逆转录获得的cDNA上杂交一个含有DNA标签(或带有几个碱基缺口)的锁式探针,并利用DNA聚合和连接反应填充间隙,通过RCA反应在细胞内产生高密度的DNA纳米球;利用标记有独特荧光的核酸探针与待测序列杂交,通过边连接边测序策略读取DNA标签(或待测转录本)的具体序列[65]。2016年Cartana公司正式将该技术商品化。该方法虽首次实现了待测转录本的原位测序,但仅对靶标位点的已知序列进行测定。Church课题组发展的FISSEQ技术,利用随机引物产生cDNA后通过单链DNA环化酶对cDNA进行环化,经RCA信号放大后对转录本本身进行测序以获取转录本序列信息,从而实现对未知靶标的测量[66,76]。然而FISSEQ的测序结果中rRNA含量高(40%~80%),可测得的mRNA种类少(~200种);同时FISSEQ产生的读长较短(5~30 bp),难以充分揭示单细胞间基因表达的差异。为了增加测序读长,同时提高成像分辨率,Church课题组进一步发展了Exseq技术(图3B)[67]。该技术通过增加一轮非原位的经典二代测序获得全长的转录组信息,同时与原位测序数据(作为空间码)进行匹配,成功获得长读长转录组的空间位置信息;此外,Exseq结合扩展显微镜技术将生物大分子、小分子等交联到可吸水放大的丙烯酰胺凝胶上,通过组织吸水放大(~4倍放大)减少了成像信号的堆积干扰,实现了纳米尺度的RNA原位测序分析。整体而言,尽管荧光原位测序在对未知靶标的测量及在空间分辨率上具有优势,但其转录本检测效率低、测序读长短、图像处理复杂等问题仍有待进一步解决,同时需要整合算法分析寻找细胞边界以确定RNA分子归属的细胞。
3.4 原位捕获技术
原位捕获技术的核心是利用具有空间位置信息的DNA标签引物捕获并标记转录本的空间位置。近几年原位捕获技术发展迅猛,并朝着提高空间分辨率及空间多组学联合分析两个方向发展。2016年Ståhl课题组首次提出了原位捕获技术(ST)[68]。ST将含有空间编码、分子编码的mRNA特异捕获引物簇固定于载玻片表面以制备空间编码玻片;接着将组织切片贴于空间编码玻片,并对其进行固定、染色与成像;利用玻片上的引物捕获经透化后的组织释放的mRNA,并通过逆转录过程生成含有空间编码的cDNA,经转录组文库制备、高通量测序与分析,将转录组序列映射至原空间位置,实现高覆盖度的组织空间转录组测量分析。之后,ST技术被成功转化为商品化平台(10×Genomics Visium)。虽然该方法分析面积大(6.5 mm×6.5 mm),但需要将大量DNA空间编码探针固定于载玻片表面,修饰成本高,且空间分辨率有限(55 μm/编码点阵),难以达到单细胞水平。为了进一步提高空间分辨率,Rodriques等用编码微球代替ST的空间点阵列,发展了Slide-seq技术[69]。Slide-seq将空间编码、分子编码与捕获序列修饰于编码微球上,并将微球紧密堆积在玻璃一侧形成单层结构。每个微球的空间编码不同,可采用SOLiD技术进行空间编码的解码。由于微球的直径为10 μm,与单个细胞的尺寸相近,因此Slide-seq方法的空间分辨率可达10 μm。Vickovic等[70]发展的高分辨率空间转录组(HDST)通过使用2 μm编码微球并结合顺序杂交策略进行微球解码,将分辨率提高至2 μm水平。李俊熙课题组发展了Seq-scope技术,基于Illumina测序芯片构建空间编码阵列后,对组织的空间转录组学进行解析,将空间分辨率提高至1 μm水平[71]。我国华大基因也推出了Stereo-seq技术,利用自组装的DNA纳米球制备空间编码阵列,进一步将空间分辨率提高至220 nm,并且可分析的组织面积达到42.25 cm2,远高于其他技术[72]。
在空间多组学研究方面,樊荣课题组发展的DBiT-seq技术开创了先河(图3C)[73]。不同于利用空间阵列或微(纳)球捕获释放的mRNA,DBiT-seq利用微流控技术对组织细胞进行原位的空间编码标记,实现了空间转录组和蛋白质的联合测量。以甲醛固定的组织玻片为起始材料,将其与识别目标蛋白的DNA标签修饰抗体(ADT)混合物共孵育;接着将含有50个平行微通道的微流体芯片放置在组织玻片上,以引入第一组DNA条码(A1~A50),其通过oligo-dT结合mRNA或ADT的poly(A)尾部;原位逆转录后,将与第一个芯片通道垂直的微流控芯片放置在组织载玻片上,以引入第二个DNA条形码(B1~B50),使其与含有A条形码的cDNA连接,从而生成AiBj(i=1~50,j=1~50)数组。从组织中提取cDNA,并通过PCR扩增制备测序文库,通过识别cDNA及抗体DNA标签上连接的空间编码AiBj重建空间表达图。依照类似的方法,该课题组进一步将该方法拓展至空间分辨的染色质可及性(spatial-ATAC-seq)及组蛋白修饰(Spatial-CUT&Tag)研究,有望实现表观遗传学、转录组、蛋白质等多种组学的联合分析[74-75]。

图3 基于调控核酸杂交热力学的DNA序列编码的荧光标记技术(SeqEA)[64](A);纳米级分辨率的完整组织RNA原位测序技术(Exseq)[67](B);基于确定性标记的空间转录组和蛋白质联合测序技术(DBiT-seq)[73](C)Fig.3 DNA-sequence-encoded rolling circle amplicon for single-cell RNA imaging based on nucleic acid hybridization regulation(SeqEA)[64](A);in situ RNA sequencing in intact biological systems with nanoliter resolution(Exseq)[67](B);spatially multi-omics sequencing via deterministic barcoding in tissue(DBiT-seq)[73](C)
本文在表1中对各类方法进行了直接对比。

表1 空间转录组分析方法对比Table 1 Comparison of spatial transcriptome analysis methods
综上所述,原位捕获技术因具有分析通量高、灵敏度高、可分析的组织面积大、可进行多模态分析等优点,成为单细胞空间分辨分析的重要技术。然而,尽管当前的原位捕获技术已实现纳米级分辨率,但基于微球/纳米球的编码方法需利用高通量测序仪对芯片上每一个编码位置进行从头测序以获得每个地理坐标位置的编码序列,过程复杂、耗时长、成本高,限制了其大规模应用;同时亚细胞水平的空间转录本定位研究仍然受限。开发制备易、通量高、成本低、面积大、灵敏度高、分辨率高的原位捕获技术并进行应用拓展是这一领域的未来发展方向。
4 结论与展望
本文综述了单细胞和空间转录组分析的前沿进展,总结并讨论了单细胞转录组测序、单细胞多组学测序和空间转录组分析方法。可以看到,当前的单细胞测序技术正朝着空间化与多组学化方向发展。尽管目前单细胞多组学及空间转录组分析在揭示单细胞异质性及细胞间的相互作用方面取得了重要突破,但仍面临若干重大挑战:首先,用于样品制备或测序的自动化设备成本较高,且需要权衡细胞数量和测序深度以控制成本;第二,大多数单细胞转录组研究局限于poly(A)尾mRNA分析,而鲜有非多聚腺苷酸RNA(如非编码RNA、小RNA等)的联合分析,亟需发展针对全转录组的单细胞空间多组学研究方法;第三,目前基于原位捕获的空间转录组学研究虽拥有了较高的基因测量数,但单细胞或亚细胞的空间分辨率有限,而基于成像的空间转录组学研究虽拥有纳米级分辨率,但基因检测数目有限,兼具高基因检测通量和高空间分辨率的分析方法是未来的发展方向;第四,当前的空间转录组分析局限于二维空间水平,尽管已有一些课题组利用连续切片组织进行二维空间基因表达分析,然后采用算法分析重构三维图谱[77],但这种方法仍存在过程复杂、切片质量无法保证等问题,完美的三维结构重塑仍未解决。因此,开发低成本、全方位、高通量、高分辨的单细胞空间多组学分析方法,实现对单个细胞更为细致、全面及准确的描述和精准单细胞图谱构建,将更具研究及应用潜力。
