A tandem asymmetric oxidation-oxa-Michael sequence for dearomatization of β-naphthols
2022-09-15LinqingWangHaiyongZhuTianyuPengYingfanXuYanzheHouShixinLiShimingPangHailongZhangDongxuYang
Linqing Wang, Haiyong Zhu, Tianyu Peng, Yingfan Xu, Yanzhe Hou, Shixin Li,Shiming Pang, Hailong Zhang, Dongxu Yang
Key Laboratory of Preclinical Study for New Drugs of Gansu Province, Institute of Drug Design & Synthesis, School of Basic Medical Sciences, Lanzhou University, Lanzhou 730000, China
ABSTRACT A catalytic asymmetric hydroxylative dearomatization reaction has been disclosed, and the products can smoothly transform into spiroannulation adducts by simply treated with a base under mild conditions.Novel in-situ generated magnesium catalytic methods are developed by application of combinational ligands.Related concise transformaitons of the spiroannulation adducts have been carried out.
Keywords:Oxidative dearomatization Oxa-Michael reaction Asymmetric reaction Chiral ligands Magnesium catalyst
Catalytic asymmetric dearomatization (CADA) reaction is a highly attractive protocol in recent years for chemists, because it is a straightforward strategy to effectively build three-dimensional chemical structures from aromatic compounds [1–11].In this context, the dearomatizative spiroannulation reactions of phenols are also rapidly developed for it’s powerful ability on construction of spirocyclic molecules.There are several representative methods for dearomatizative spiroannulation reactions, for example, one is the intramolecular pathway, including oxidation and substitution reactions [12–21]; another pathway can be intermolecular cyclization reactions, such as the catalytic asymmetric [1+4] and [2+3]spiroannulations reported very recently [22–25].
The catalytic asymmetric oxidative dearomatization reaction of phenols can break the aromaticity to establish the stereochemical structure by simply equipping a hydroxyl group to the molecules.Several catalytic methods, for example, hypervalent iodine(iii) reagent [12–17], diamine-copper [26] orN,N’-dioxide–scandium catalysts [27] have been successfully developed for the asymmetric hydroxylative dearomatization of phenol compounds.However, the generated chiral tertiary alcohols have less been utilized for further intramolecular additions to construct more complicated skeletons [28], such as oxaspirocyclic molecules.Herein,on the basis of recent works on asymmetric dearomatization reactions of phenol compounds [29–32], we designed a tandem asymmetric oxidation-oxa-Michael reaction sequence for dearomatization ofβ-naphthols 1, by developingin-situgenerated magnesium catalytic methods with combinational use of different ligands [33–51].The current reaction including a Mg(II)-catalyzed enantioselective oxidation step to generate the hydroxylation adduct 2 and a base promoted intramolecular oxa-addition step to finish the formal spiroannulation process (Scheme 1) [52–56].
Scheme 1.The designed tandem asymmetric oxidation-oxa-Michael reaction.
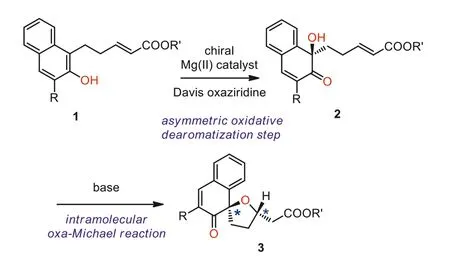
Scheme 2.Initial optimization process of the tandem asymmetric oxidationoxa-Michael reaction.Reactions were performed with naphthol 1a (0.10 mmol),4 (0.10 mmol) in toluene (1.0 mL) in the presence of Bu2Mg (20 mol%) and L4(20 mol%).
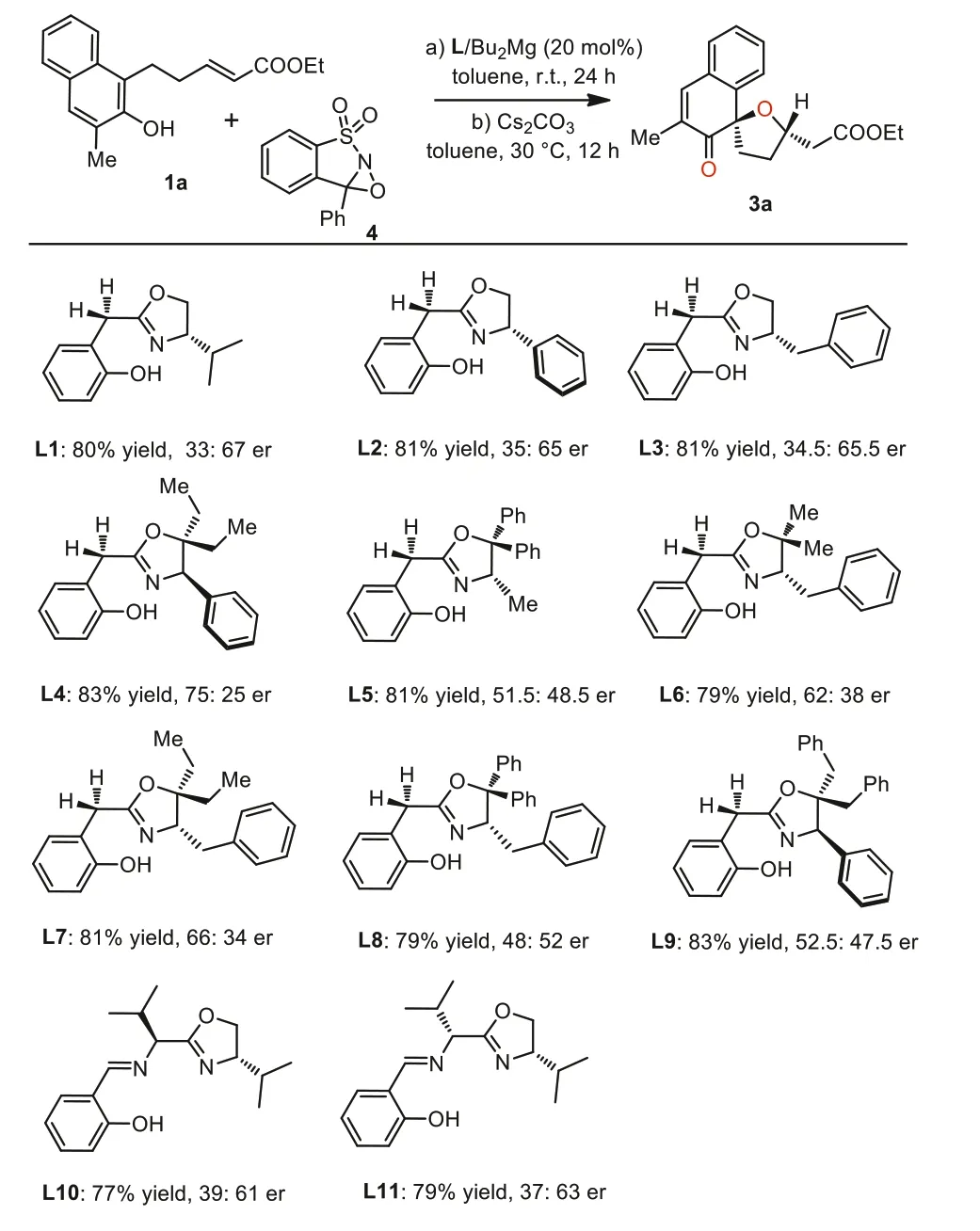
In the initial experiment of the tandem asymmetric oxidationoxa-Michael reaction between naphthol derivative 1a and Davis oxaziridine 4 [57,58], varies ofin-situgenerated magnesium catalysts employing of different chiral ligands were examined, and Cs2CO3was identified as the optimal base for the stepwise intramolecular oxa-Michael reaction, resulting in good yields and diastereoselectivities (>20:1 dr).The employment of BINOLs or Salen as chiral ligands revealed that these magnesium catalysts could mediate the dearomatizative hydroxylation reaction of naphthol 1a, and afforded the oxidative dearomatization product 3a in excellent yields but with disappointed enantioselectivities.The detail optimization process is deposited in Supporting information.Further evaluation of oxazoline-OH ligands with different substituents and backbones indicated that all the reactions proceeded smoothly to form the desired dearomative product 3a with variable enantioselective control (Scheme 2).We found the ligands with different chiral substituents gave similar results (Scheme 2, L1–L3),some poly-substituted ligands were also introduced and led to dramatically different enantioselectivities.The screening progress finally identified L4 as a more promising chiral ligand, which was derived from a multi-substituted amino alcohol, leading to the oxidative dearomatization product 3a in an excellent yield with a moderate enantioselectivity (83% yield, 75:25 er).
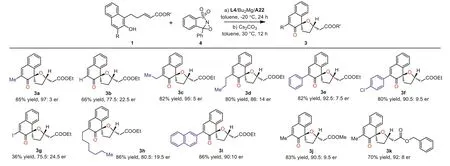
Scheme 3.Substrate scope of the reaction.Reactions were performed with naphthol 1a (0.20 mmol), 4 (0.20 mmol) in toluene (2.0 mL) in the presence of Bu2Mg (20 mol%)and L4 (30 mol%) and the second ligand A22 (40 mol%).
To further improve the enantioselectivities of this oxidative dearomatization reaction, we turned to introduce a second ligand and tested its effects on the magnesium catalysis.As illustrated in Table 1, a wide scope of additives were examined as the second ligands in the Mg(II) mediated dearomatization process.The introduction of phenol such as A1 obviously decreased er value (Table 1, entry 1).The introduction of phosphamide or sulfamide did not show promise results (Table 1, entries 2 and 3).Utilization of various of chiral sulfonamide obviously improved the er values, and we found some benzamides could also increase the enantioselectivities in the presence of L4/Bu2Mg, and the use of 1-naphthamide A16 gave a relatively better enantioselectivity but with slightly lower yield.To our delight, when oxazolidone A20 was employed as theadditive ligand, the er value of the model reaction increased obviously to 83.5:16.5 (Table 1, entry 20).Encouraged by this result,a series of optically substituted oxazolidones were further examined and the ligand A22 led to relatively better results.Interestingly, it was found that the addition of (R)-oxazolidone A22 resulted in slight higher er value than its optical isomer A21, and the increase of steric hindrance of the substituents on (R)-oxazolidones would lead to decreased er values of the oxidative dearomatization product 3a (Table 1, entries 23–25).Further increasing the loading amount of L4 and A22 led to the optimized results of the tandem asymmetric oxidation-oxa-Michael reaction.
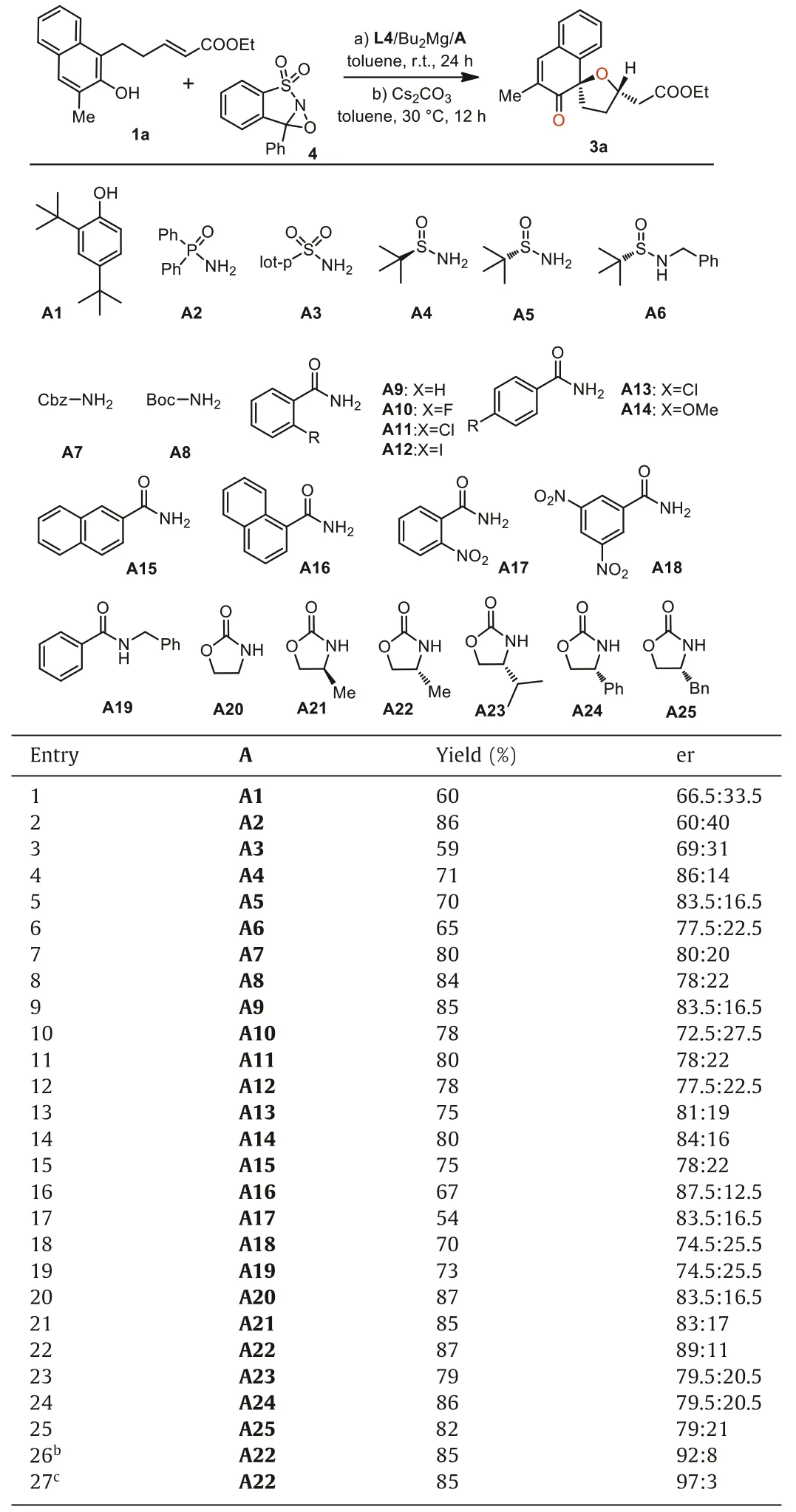
Table 1 The optimization process of combinational magnesium catalyst for the reaction.a
With the development of suitable conditions and identification of combinational magnesium catalyst generating from L4, A22 and Bu2Mg, we next began to investigate the substrate scope of this tandem asymmetric oxidation-oxa-Michael reaction.As the results summarized in Scheme 3, different alkyl substituted (Me, Et,n-Pr andn-heptyl)β-naphthols were well tolerated, and their corresponding dearomative products were obtained in excellent yields and good enantioselectivities (3a, 3c, 3d and 3h).β-Naphthols bearing aryl substituents at C3 position, challenging substrates in some intermolecular asymmetric oxidative dearomatization reactions, were also worked well and led to the desired spirocyclic hydrofuran structures in good yields and enantioselectivities (3e,3f, 3i).Moreover, naphthols with halogen or hydrogen atom at the C3 position, which were often failure substrates in dearomatization reactions of phenol compounds, were also tolerable but resulted in relatively lower enantioselectivities (3b, 3g).Furthermore, two other substrates with different unsaturated esters equipped at the C1 position also underwent the dearomative reactions smoothly with satisfied enantioselectivities (3j, 3k).
Next, several transformations of the spiroannulation adducts were carried out (Scheme 4).The ester group and carbonyl group in the cyclohexanone frameworks of 3a could be readily reduced by lithium aluminum hydride to the diol product 5 in excellent yield and diastereoselctivites, and the terminal alcohol was selectively protected withp-toluene sulfonyl group by simply treated with tosyl chloride and triethylamine in the presence of DMAP, led to the compound 6, and the absolute structure was analyzed by corresponding X-ray crystallographic studies.Moreover, compound 7 with an oxygen bridged ring was obtained in a high yield by simply treating 5 with SOCl2at mild conditions.
On the basis of our previous works on developing ofin-situgenerated magnesium catalysts with the utilization of oxazoline-OH chiral ligands [29,31,46,48], a hypothesized mechanism of this Mg(II)-catalyzed hydroxylative reaction is proposed (Scheme 5).The magnesium compound I was confirmed in our former works and we speculated the bidentate coordinate phenol 1 is prone to finish another neutralization step to generate the complex II.The oxaziridine might coordinate to the magnesium centre in the less steric hindered direction and the oxazolidone A22 can occupy another possible coordination site, which might be the reason to improve the enantioselectivities (III).Then the hydroxylative step occur at the favoredSi-face to generate the dearomatization product.Next release the adduct 2 and the generated imine initiate another catalytic cycle after replacement with the bidentate phenol 1 and another molecule of oxaziridine.
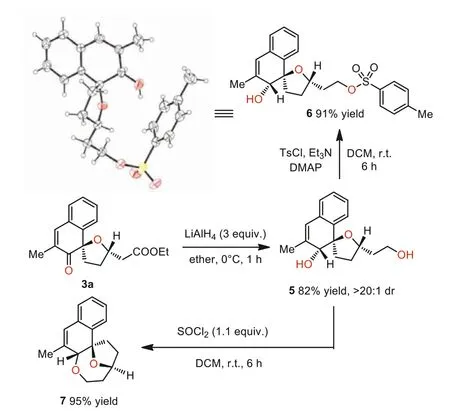
Scheme 4.Transformation of the formal spiroannulation product.
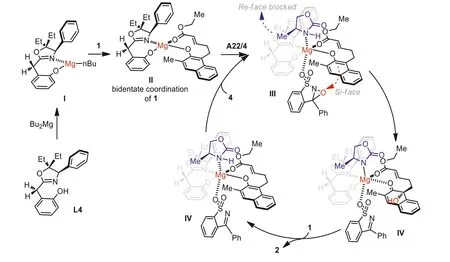
Scheme 5.A hypothesized mechanism of the asymmetric hydroxylative dearomatization reaction.
In summary, we have developed a combinational magnesium catalytic method for the tandem asymmetric dearomatizative oxidation-oxa-Michael reaction of the naphthol compounds.The introduction of additives such as oxazolidones can obviously improve the enantioselectivities.Representative substituents on the naphthols were examined and led to satisfied results in this reaction.And several brief transformations of the spiroannulaiton adducts were carried out in good results.Our future work will focus on developing ofin-situgenerated magnesium catalysts and applying them in more catalytic asymmetric reactions.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We acknowledge the financial support from the National Natural Science Foundation of China (Nos.21901092, 21807053), Innovation Fund for Medical Sciences (No.2019-12M-5-074), Program for Chang-jiang Scholars and Innovative Research Team in University (PCSIRT) (No.IRT_15R27), the Funds for Fundamental Research Creative Groups of Gansu Province (No.20JR5RA310), and the Fundamental Research Funds for the Central Universities (Nos.lzujbky-2020-49, 2021-kb21).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.12.075.
杂志排行

Chinese Chemical Letters的其它文章
- A review on recent advances in hydrogen peroxide electrochemical sensors for applications in cell detection
- Rational design of nanocarriers for mitochondria-targeted drug delivery
- Emerging landscapes of nanosystems based on pre-metastatic microenvironment for cancer theranostics
- Radiotherapy assisted with biomaterials to trigger antitumor immunity
- Development of environment-insensitive and highly emissive BODIPYs via installation of N,N’-dialkylsubstituted amide at meso position
- Programmed polymersomes with spatio-temporal delivery of antigen and dual-adjuvants for efficient dendritic cells-based cancer immunotherapy