NFSI-catalyzed S–S bond exchange reaction for the synthesis of unsymmetrical disulfides
2022-09-15MengjieSongQingyueHuZhengYiLiXioqingSunKeYng
Mengjie Song, Qingyue Hu, Zheng-Yi Li,∗, Xioqing Sun, Ke Yng,∗
a Jiangsu Key Laboratory of Advanced Catalytic Materials & Technology, School of Petrochemical Engineering, Changzhou University, Changzhou 213164,China
b Institute of Urban & Rural Mining, Changzhou University, Changzhou 213164, China
ABSTRACT The metal-free S–S bond exchange reaction of symmetrical disulfides catalyzed by NFSI is described.This novel protocol provides a facile and efficient approach to accessing important unsymmetrical disulfides.Furthermore, this strategy could also be utilized in the late-stage functionalization of amino acids, drugs,and natural products.The broad substrate scope, good functional group tolerance and easy accessibility of catalyst indicate that this strategy affords a green and practical complementary method to various unsymmetrical disulfides.
Keywords:Metal-free S–S bond exchange NFSI Unsymmetrical disulfides Organic synthesis
Unsymmetrical disulfides, as important organic sulfurcontaining motifs, are widely present in drugs and natural products (Fig.1a) [1–4].Furthermore, unsymmetrical disulfides also play a vital role in life science [5].Due to their importance in different fields, the synthesis of unsymmetrical disulfides has received extensive attention.Previous reports on the construction of such skeletons are divided into two categories: one is S–S bond formation [6–25] and the other is C–S bond formation [26–33].The S–S bond-forming methods (Fig.1b) usually include the direct oxidative dehydrogenation coupling [6–14], SN2 replacement[15–20], and exchange reaction [21–25].While the C–S bond formation strategy (Fig.1c) generally involves the cross-coupling or nucleophilic substitution of masked disulfuration reagents[26–32], and the radical substitution of tetrasulfides [33].Among these, disulfide exchange reaction has been regarded as one of the most direct and effective approaches to construct unsymmetrical disulfides [21–25].Furthermore, disulfide exchange process, one of the most popular dynamic covalent chemistries, has also been widely applied in biological and material systems [34,35].Some early pioneering works by utilizing Rh catalysts have proven to be successfully.Yamaguchi group developed a Rh-catalyzed disulfide exchange strategy to access unsymmetrical disulfides [21,22].Since then, only a few methodologies on disulfide exchange process have been reported [23–25].Therefore, the development of a facile, efficient and green method to access a variety of unsymmetrical disulfides is still challenging [36].
In the past decade,N-fluorobenzene sulfonimide (NFSI) has attracted much attention due to its widely applications in fluorination and amination reactions [37,38].However, the use of NFSI as the organic catalyst to achieve the disulfide metathesis has not been reported.In our continuing efforts for developing novel strategies for sulfur-containing compounds [39–46], herein, we disclosed a NFSI-catalyzed S–S bond exchange reaction to access various important unsymmetrical disulfide derivatives (Fig.1d).
The investigation began with the disulfide exchange reaction between 1,2-bis(4-methoxyphenyl)disulfane 1a and dimethyl disulfide 2a in the presence of NFSI (10 mol%) at 50°C (Table 1).To our delight, the initial reaction solvent screening indicated that DCE(1,2-dichloroethane) was the optimal solvent, providing the desired product 3a in 83% isolated yield (Table 1, entries 1–7).The subsequent examination of different F+reagents, including Selectfluor and NFPT, showed that none of other F+reagents provided better results than NFSI (Table 1, entries 8 and 9).It should be noticed that the yield of desired product 3a could not be improved any more by increasing or decreasing the amounts of NFSI (Table 1, entries 10–12).Furthermore, this disulfide exchange process at room temperature (25°C) also provided the desired product 3a in 61% isolated yield (Table 1, entry 13).Finally, only trace of product 3a could be detected when NFSI was absent (Table 1, entry 14).
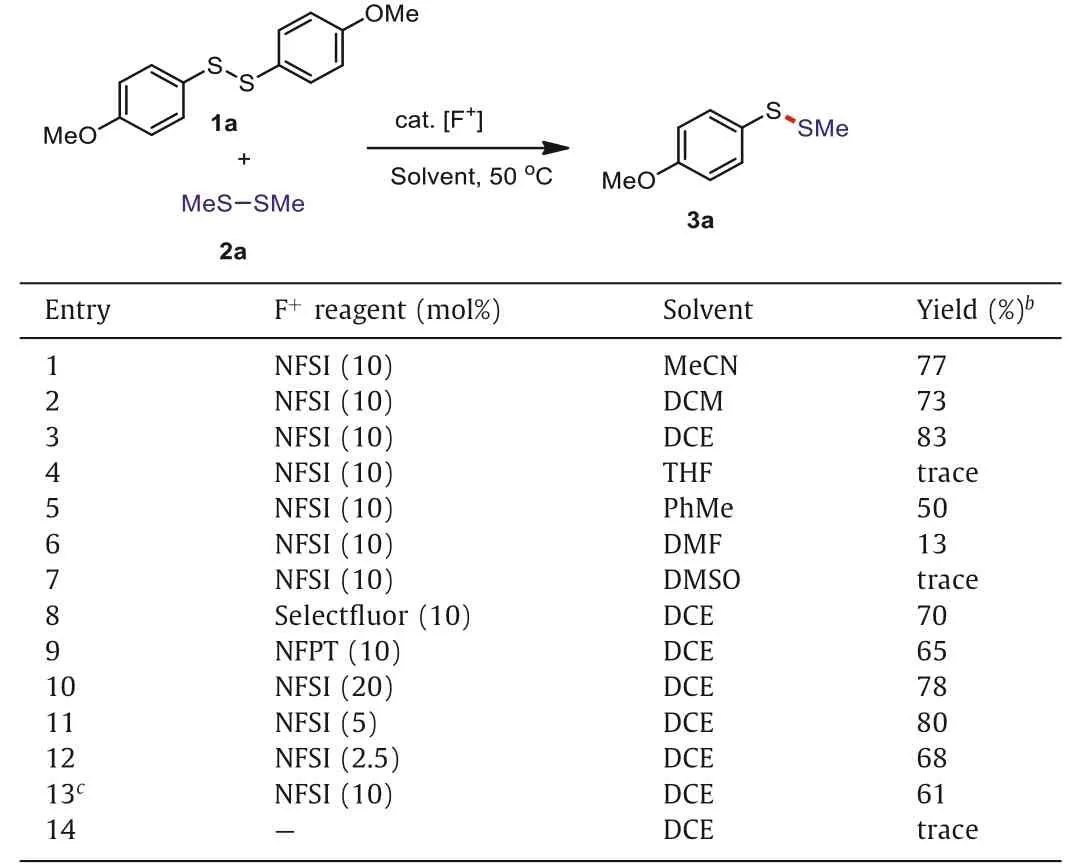
Table 1 Optimization of reaction conditions.a
With the optimized reaction conditions in hand, we explored the exchange reaction between diaryl and dialkyl disulfide.Asshown in Scheme 1, the dialkyl disulfides bearing different substituent groups, including methyl, ethyl,n-Pr,n-Bu,i-Pr, cyclopentyl and benzyl, were smoothly converted to the corresponding products 3a–3g in good yields.Reactions of dimethyl disulfide with different di(hetero)aryl disulfides also generated the desired products 3h–3k in good yields.Furthermore, two different hindered disulfides afforded the desired product 3l in 35% yield.In addition, two different kinds of symmetrical dialkyl disulfides were used to exchange with each other, providing unsymmetrical dialkyl disulfides 3m–3r in moderate to good yields.
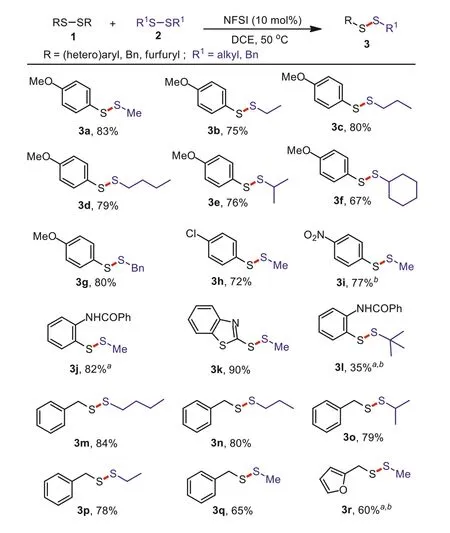
Scheme 1.Substrate scope for unsymmetrical aryl-alkyl and dialkyl disulfides.Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), NFSI (0.02 mmol), DCE (2.0 mL), 50°C,4 h.Isolated yields (based on 0.4 mmol of 3), aNFSI (0.04 mmol). b80°C.
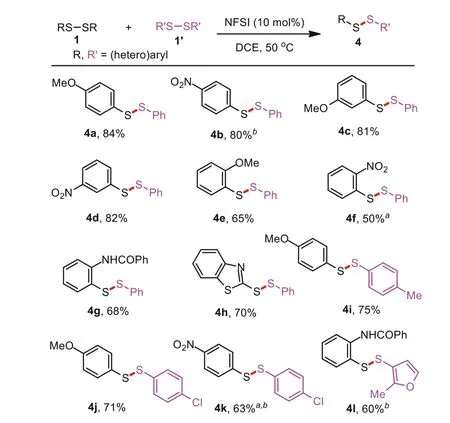
Scheme 2.Substrate scope for unsymmetrical diaryl disulfides.Reaction conditions: 1 (0.2 mmol), 1′ (0.4 mmol), NFSI (0.02 mmol), DCE (2.0 mL), 50°C, 4 h.Isolated yields (based on 0.4 mmol of 4). aNFSI (0.04 mmol). b80°C.
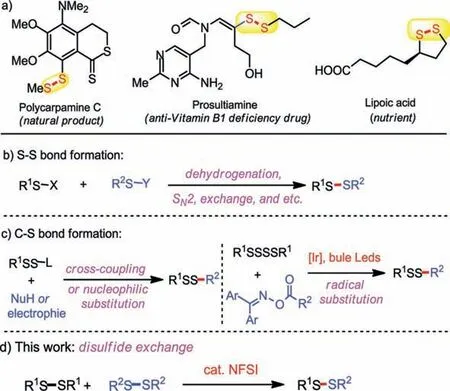
Fig.1.Selected important unsymmetrical disulfide derivatives and strategies for unsymmetrical disulfide derivatives.
The substrate scope of previous reports mainly focused on preparing unsymmetrical dialkyl or aryl-alkyl disulfides [21,22].In this protocol, the generality of the exchange reaction for preparing different unsymmetrical diaryl disulfides was carefully examined in Scheme 2.Diaryl disulfides 1 bearing electron-withdrawing or electron-donating groups at thepara-,meta-andortho-position on the phenyl ring were well compatible with PhSSPh under the current catalytic conditions, affording the desired products 4a–3g in moderate to good yields.Next, the exchange reaction of 1,2-bis(benzo[d]thiazol-2-yl)disulfane and diphenyl disulfide also led to the corresponding product 4h in 70% yield.In addition, diaryl disulfides 1′with different substituent groups (Me and Cl)smoothly coupled with 1,2-bis(4-methoxyphenyl)disulfane to produce the desired products 4i and 4j in good yields.Furthermore,two substrates with different electron-withdrawing groups (NO2and Cl) also generated the desired product 4k in 63% yield.Notably, the desired product 4l was isolated in 60% yield, indicating that the steric hindrance of aryl disulfides has a slight effect on this catalytic system.
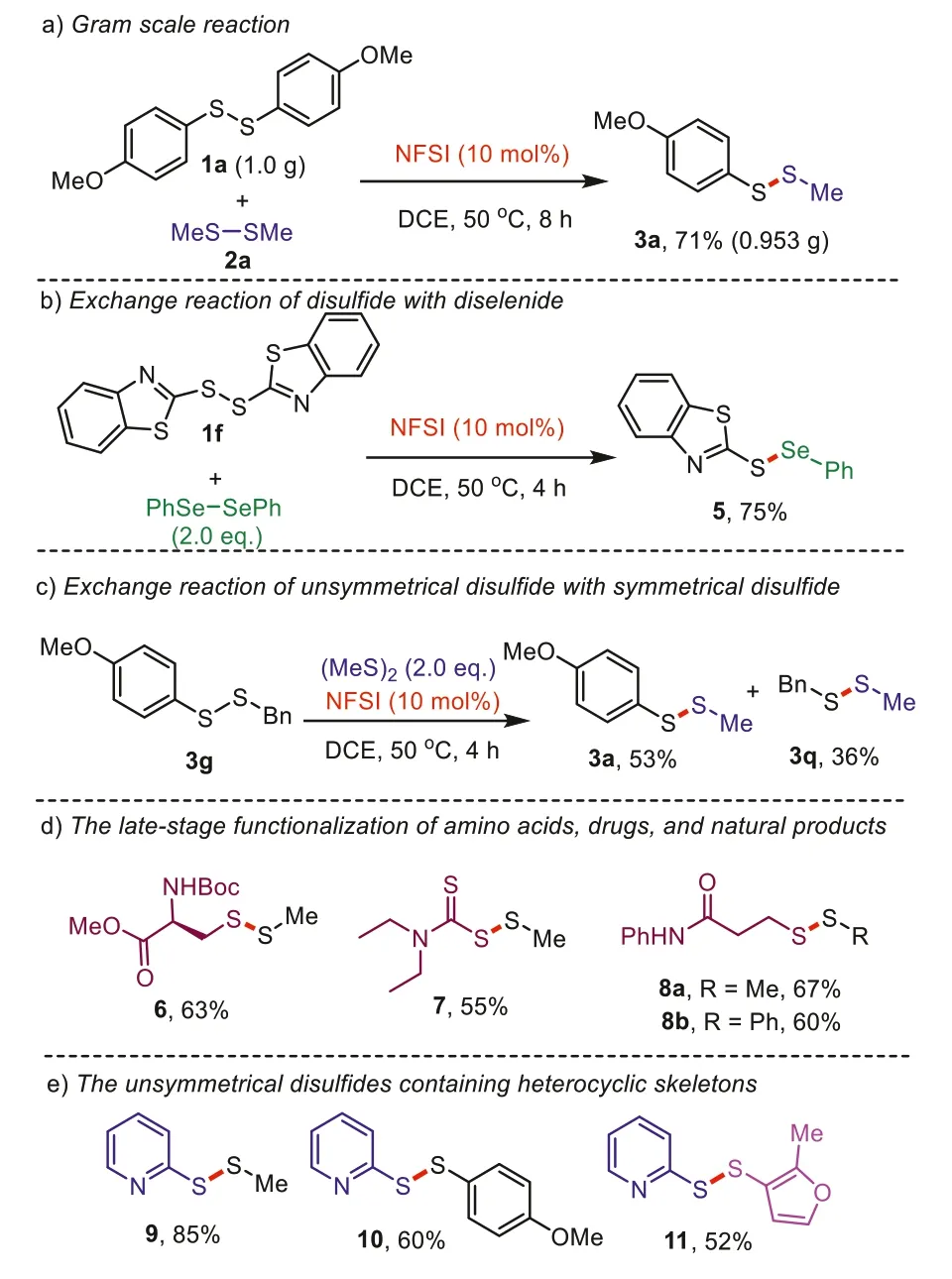
Scheme 3.Further exploration of the reaction scope.
To study the synthetic utility of this catalytic system, further exploration of the reaction scope was carried out in Scheme 3.A gram-scale reaction was performed to afford the product 3a with 71% isolated yield (Scheme 3a).Furthermore, the exchange reaction between 1f and (PhSe)2provided the desired product 5 in 75%yield (Scheme 3b).Next, unsymmetrical disulfide 3g could react with (MeS)2to produce two different unsymmetrical disulfides 3a and 3q in 53% and 36% yields, indicating that the aryl-alkyl disulfide formation may be preferred than alkyl-alkyl disulfide formation (Scheme 3c).To our delight, this protocol could also be utilized in the late-stage functionalization of amino acids, drugs, and natural products (Scheme 3d).Amino acid derivatives (Boc-L-Cys-OMe)2produced the desired product 6 in 63% yield.The disulfiram drug could be functionalized to synthesize the product 7 in moderate yield.In addition, dithiodipropionamides, as important intermediates for the synthesis of biological activity isothiazolone derivatives, were transformed to the corresponding products 8a and 8b in 67% and 60% yields, respectively.Finally, both unsymmetrical disulfides containing pyridine moieties 9,10 and bishetroaryl disulfide 11 were prepared in moderate to good yields under the optimized reaction conditions.
To obtain some insights into this reaction, some control experiments have been carried out (Scheme 4).The radical trapping experiment was performed and this result suggests that a single electron transfer (SET) may not be involved in this catalytic process(Scheme 4a).In addition, substrate proportion control experiments between 1a and 2f indicate that an excess of one partner can enhance the efficiency of reaction (Scheme 4b).Intermolecular competition results between 1a and 1d reveal that the reaction rate of electron-rich substrate is superior to electron-deficient substrate(Scheme 4c).
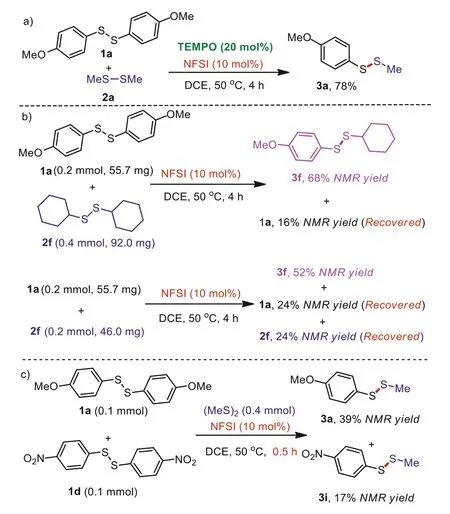
Scheme 4.The control experiments.
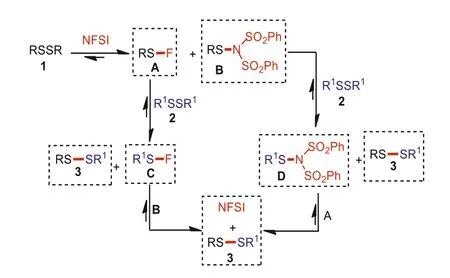
Scheme 5.The plausible reaction mechanism.
Based on the control experiments and previous reports[23,24,47–53], a plausible catalytic cycle is depicted in Scheme 5.It is believed that this catalytic cycle is a reversible process in the presence of NFSI.First, treatment of RSSR 1 with a catalytic amount of NFSI catalyst generates the intermediate A and B.Then, the subsequent exchange reaction between intermediate A and R1SSR12 affords the intermediate C and desired unsymmetrical disulfide 3.Meanwhile, the intermediate B can also react with R1SSR12 to provide intermediate D and product 3.Finally, the reaction of intermediate B and C (or intermediate D and A) produces the desired product 3 and regenerates the NFSI catalyst again.It is worth mentioning that this process can also be initiated from the reaction of NFSI with R1SSR12.
In summary, we have demonstrated a novel disulfide exchange reaction for the efficient construction of unsymmetrical disulfides by employing NFSI as the catalyst.To the best of our knowledge,this is the first NFSI-catalyzed disulfide exchange process.Moreover, this green strategy also provides a complementary approach to diverse unsymmetrical disulfides, which of them are important skeletons of amino acids, drugs and natural products.Further study on the reaction mechanism and application is ongoing in our laboratories.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Nos.21572026,21702019), Postgraduate Research & Practice Innovation Program of Jiangsu Province (No.SJCX20_0952), Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, Changzhou University.We also acknowledge the analytical testing support from Analysis and Testing Center, NERC Biomass of Changzhou University.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.12.073.
杂志排行

Chinese Chemical Letters的其它文章
- A review on recent advances in hydrogen peroxide electrochemical sensors for applications in cell detection
- Rational design of nanocarriers for mitochondria-targeted drug delivery
- Emerging landscapes of nanosystems based on pre-metastatic microenvironment for cancer theranostics
- Radiotherapy assisted with biomaterials to trigger antitumor immunity
- Development of environment-insensitive and highly emissive BODIPYs via installation of N,N’-dialkylsubstituted amide at meso position
- Programmed polymersomes with spatio-temporal delivery of antigen and dual-adjuvants for efficient dendritic cells-based cancer immunotherapy