药物中间体合成工艺的研究
2022-09-14王效杰漆定超张宝成
*王效杰 漆定超 张宝成
(1.沧州那瑞化学科技有限公司 河北 061100 2.河北省糖尿病和精神疾病药物中间体重点实验室 河北 061100)
引言
近年来,药物生产技术水平的日益提升,将中间体作为原料的药物种类数量也逐渐增多,药物中间体行业发展规模逐渐壮大。但与此同时,在研发药物中间体时,对工艺以及技术要求越来越高,必须保证工艺路线制定的合理化及科学化,以此让中间体能高效合成。为实现这一目标,需要加强分析中间体合成工艺,不断对传统合成工艺技术优化和创新,增强合成工艺操作简便性。
1.药物中间体研发需求
药物生产期间所得的中间产品被称为药物中间体,可以作为辅料或者原料,与一般的药品存在较大差异,药物中间体属于化工原料或者产品,在对中间体生产期间,即便没有药品生产许可证也能完成,石油化工厂便能对药物中间体进行生产。药物中间体的用途专一,能让整个生产过程实现精细化目标[1]。现阶段,市场中出现的药物中间体类型较多,包括维生素类中间体等,可以从中获得很高利润。很多化工企业在经营过程中,为了能促进自身经济效益的提升,对药物中间体的生产越来越重视。从整体角度分析,随着工艺技术水平的不断提升,药物中间体的合成工艺也越来越成熟,实现了大规模生产,但和国外药物中间体相比,我国药物中间体的附加值仍有很大提升空间,再加上市场价格的不断提升,很多药物中间体的研发受到局限,因此要加强重视,深入对合成技术研究和优化[2]。
2.药物中间体合成工艺
(1)药物中间体合成工艺简述。在合成药物中间体过程中,所采用的途径较多,而具有工业生产价值的合成方法则被称为工艺。在实际合成药物中间体期间,由于选择的原料以及合成步骤流程存在差异,应用的工艺路线也会有很大区别。在对新药研制过程中,应该在实验室开展化学合成,明确性质优异后,加强对工艺的研究和分析,依照生产规模,有针对性地利用合成工艺,保证合成的中间体能达到最优,减少资源的消耗,尽可能让生产成本降到最小[3]。现阶段,药物中间体的合成工艺主要分为两种,一种为全合成工艺,另一种为半合成工艺。其中,全合成工艺在应用期间,主要是应用基本结构天然产物,采取化学改造和物理处理的方式,以此获得复杂化合物。在全合成期间,需要应用的方法简单,操作便利,不需要太过繁琐的流程便能获得复杂化合物。在对药物中间体合成期间,涉及的流程相对较多,因此要根据具体情况选择不同种类的合成技术。在对原料产物分析期间,还要加强对工艺成本的控制,明确工艺应用是否会对环境造成影响,保证所使用的合成工艺能更为合理,生产出高质量的产品。
(2)合成工艺技术。现阶段,在对药物中间体合成过程中,可以选择应用的工艺技术较多,而应用比较广泛的技术具体有以下几种:
①缩合技术。该技术具体是运用两个或两个以上有机化合物分子在相互作用和影响下形成新的大分子,释放出小分子,诸如水、醇等。在合成药物中间体过程中,通过对缩合技术的利用,能发生氨甲基化反应。氢原子被α-氨甲基取代后生成β-氨基酮类化合物。通过对缩合技术的使用,能合成杂环化合物类药物中间体,具体有氨氯地平等。在此过程中,应用的醛有甲醛等,所应用的胺具体有氨、仲胺等,可以实现酸以及碱催化[4]。
②定向硝化技术。在对药物中间体实际生产期间,通过对该技术的使用,借助载体或者非载体区域开展定向硝化操作,以此对苯体和方向稠环异构体比例进行有效调整,提升合成的效果,获得更为高质量的中间体。在硝化反应过程中,可以对中性酸合理利用,以此达到对污染全面减少的目的,保证药物中间体在合成过程中不会出现大量资源消耗问题,节约合成成本,让绿色生产要求得到满足。比如:在合成扑热息痛期间,可以利用这一技术顺利完成氨基苯酚PAP中间体合成,促进合成效率和质量的提高。
③膜分离技术。将该技术运用于药物中间体合成环节,对合成的效率有很大促进作用,能获得更高质量的中间体。该技术主要借助物理处理的方式获取化工原料或者产品。现阶段,膜分离技术在使用过程中能达到中间体浓缩的成效,产品的纯度很高。与其他技术不同,膜分离技术在实际操作阶段没有太过繁琐的流程,整个操作环节便利,能让生产成本控制在既定范围内,有利于生产厂家经济效益的提高。比如:通过对循环冷却水冷却所获得的原料液,当温度处于10~25℃时进行脱盐,最后通过使用增压泵达到增压的效果,真正达到洗盐和浓缩的目的。
3.药物中间体合成工艺的运用
在对药物中间体合成工艺分析过程中,主要将来那度胺的中间体作为研究对象。来那度胺作为一种高效的抗癌药被广泛关注,会对细胞内多种生物种产生影响,现阶段,来那度胺与其他药物的联合应用研究仍在继续。通过对来那度胺的进一步分析可知,其在市场上有很高的价值,但其暴露出来的弊端也比较多,尤其是在合成方面,现有的路线反应长,所需要花费的时间较长,同时还存在使用高毒的CCl4、环境不友好等问题。为了能让现存的工艺得到优化和改进,需要寻找绿色且温和的合成工艺,更加科学有效的合成来那度胺。在具体合成起价,应该从逆合成角层面考量,科学对合成路线加以设计,合理开展实验,探索一条既具备创新性,又具有较高实施性的路线,让以往合成路线中存在的问题能彻底解决,提升合成的效率,尽可能达到既定要求。在研究中,对传统工艺进行了优化,选择的起始原料为3-硝基邻苯二甲酸,这种材料不需要投入太大经济成本,很容易获得。
(1)实验方法
①3-硝基邻苯二甲酰亚胺的制备
结合相关的数据文献,并在综合考量收率和成本的基础上,在研究过程中,固定溶剂可以利用冰醋酸,尿素作为源胺,将反应的温度控制在117℃,反应的时间控制在3h,在经过酰胺化反应之后,对尿素的实际用量合理筛选,具体反应如下所示:
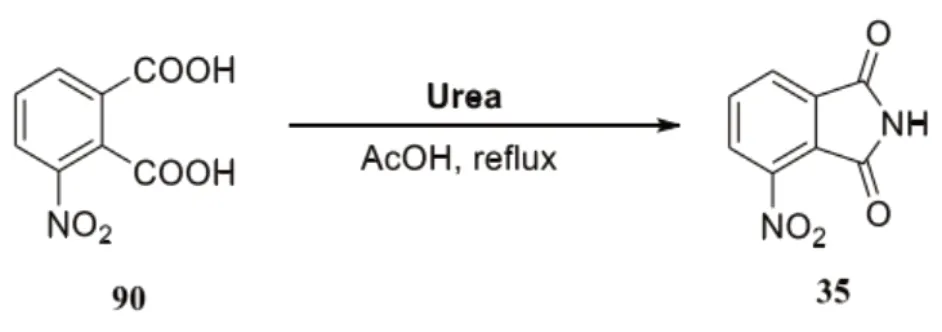
依照最终的实验可以看出,适当对尿素的投放量增加,能让目标产物35的回收率明显提升,控制化合物90的用量保持不变。逐步对尿素的涂料量进行增加,直到增加至0.9mmol,目标产物35的回收率高达98%,继续对尿素的用量进行增加,目标产物收率没有发生太大改变。通过对实验的进一步优化可知,当3-硝基邻苯二甲酸与尿素的投料比为1:0.9时,收率达到最高,具体为98%。
②4-硝基异吲哚啉的制备
运用还原羰基对吲哚啉制备过程中,应用的方法主要有两种分别,为BH3直接还原、NaBH4和BF3·Et2O先制备硼烷再还原。在本次的研究中,主要对NaBH4和BF3·Et2O先制备硼烷再还原方法加以使用,反应试剂NaBH4和BF3·Et2O,同时对反应条件加以筛选,固定反应溶剂为THF。
将THF作为溶剂,结合最终筛选出来的还原试剂,最后发现适当增加投料量能获得较高的反应收率。当NaBH4:BF3·Et2O的比例为1:10:10状态时,化合物39的收率处于最高状态,如果在此后持续增加投料,收率会呈现下降的趋势,具体反应公式如下。
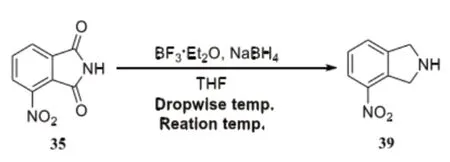
通过分析可知,对BF3·Et2O滴加温度,随着不断变化,最终能发现,温度和时间对反应收率会产生很大影响。当BF3·Et2O滴加到-20℃时,收率并没有产生太大改变。当升高滴加温度到0℃时,收率出现明显降低的情况,当升高低加温度到10℃时,收率显著下降,只有32%。所以滴加温度最佳的温度为-10℃,随后对反应温度进行了进一步观察明确,当反应温度降到45℃时,反应收率只有29%,结果表明,当反应温度为66℃,反应所获得的成效最佳,收率能达到75%。
③3-(4-硝基-1,3-二氢-2H-异吲哚-2-基)哌啶-2,6-二酮的制备
A.反应公式具体为:
通过分析表1可以看出,3-溴-2,6-哌啶二酮32,4-硝基异吲哚啉39为原料,CH3CN为溶剂,从铜盐和碱两层面考察反应添加剂。铜盐在观察一段时间后,将CuI作为添加剂,发现不添加其他添加剂,反应的收率只有32%,但如若添入其他添加剂,诸如Na2CO3、TEA,能促进反应率的提升。由此结果可以看出,CuI产生的作用较小,无法达到对反应收率整体提升的目的。与之相反,还有可能导致反应收率持续下降的情况。在实验过程中,采取不添加铜盐的方式观察碱,只添加TEA,最后的结果显示:在反应过程中,并没有改善反应收率,所获得的成效没有达到预期,但不将催化剂添加到实验中,反而会让反应收率得以提高。

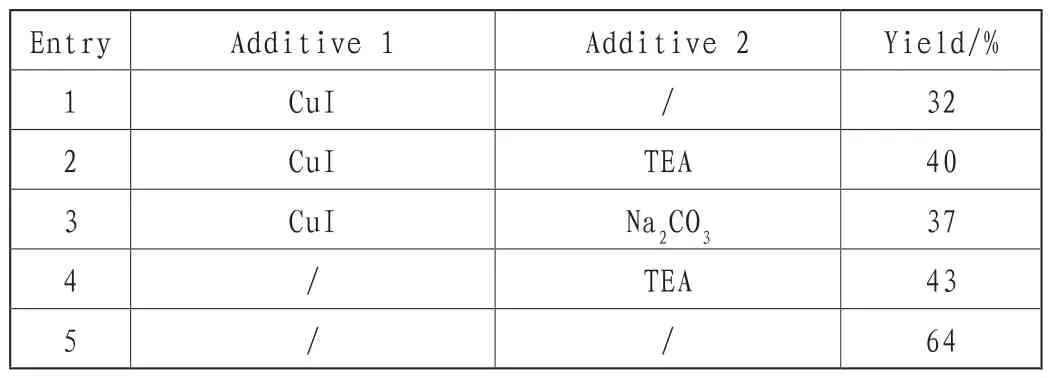
表1 添加剂对化合物40收率的影响
B.底物投料比对反应收率的影响具体公式如下:
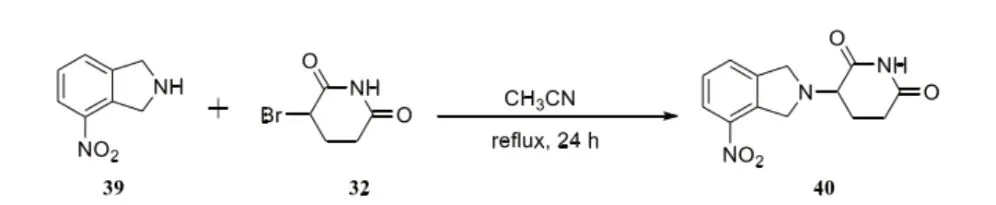
通过分析可以看出,将化合物32作为反应底物,对化合物39用量合理筛选,最终的结果显示反应收率达到49%。对化合物39的用量进行增加,当投料比为1:1.1时,收率达到55%,继续对投料量进行增加,当比例达到1:2时,收率提高到64%。随后将投料比添加到1:1.3,反应收率继续增加,具体为72%。由此在继续增加后发现收率不发生任何改变。结合结果可知,当比例为1:1.3时,收率能达到最高。
(2)实验过程
①3-硝基邻苯二甲酰亚胺的合成。在具体实验过程中,将0.211g,1.0mmol的3-硝基邻苯二甲酸,在冰醋酸溶液中溶合,冰醋酸的含量为15mL,之后将0.9mmol的尿素加入到体系中,将生成的混合物搅拌均匀,当搅拌5min后停止,继续回流反应,时间控制在3h,待整个反应过程完毕,将混合物冷却至室温,减压浓缩得粗产物。应用15mL的水和15mL的乙酸乙酯萃取3次,将残余的尿素彻底清除,在干燥有机相过程中,可以借助无水MgSO4完成,过滤并减压浓缩,最终获得0.188g黄色产物,收率达到98%。
②4-硝基异吲哚啉的合成。在进行4-硝基异吲哚啉合成实验期间,将0.192g,1mmol的3-硝基邻苯二甲酰亚胺35加入到三口反应瓶中,选择0.378g,10mmol的NaBH4,在N2的保护下加入15mL的THF,降低温度,当温度处于-10℃时,对其进行搅拌,时间控制在10min。之后利用1.25mL,10mmol的BF3·Et2O缓慢滴加,之后进行5min的保温,当温度恢复到原本的回流温度后,反应现象会持续进行,并对时间严格控制,最好为12h。反应完成后,对混合物静置一段时间,当完全冷却后,通过加入饱和NaHCO3溶液的方式,让混合物淬灭。在此过程中,速度要保持缓慢,不能速度太快,以免影响最终结果。同时运用20mL的EA稀释,使用6.0M NaOH水溶液碱化至pH10,对有机相进行分离,利用10mL的饱和食盐水洗涤,在干燥过程,通过对无水Na2SO4的合理使用,经过一段时间的过滤以及减压浓缩后,获得粗产物。为实现水相目标,在具体操作期间,采用乙醚洗涤,含量为5mL,并对盐酸加以使用,让溶液的pH值能调节到2,之后再次应用6.0M NaOH水溶液碱化至pH10,使用15mL的EA萃取,分离有机,利用饱和食盐水开展洗涤工作,干燥过程仍然选用无水Na2SO4,最终的收率最高,具体为75%。
③3-(4-硝基-1,3-二氢-2H-异吲哚-2-基)哌啶-2,6-二酮的合成。在N2的保护下,将0.123g,1.3mmol的4-硝基异吲哚啉39加入到反应瓶中,之后再往瓶中添加0.192g,1mmol的3-溴-2,6-哌啶二酮32,最后加入10mL的CH3CN,回流反应的时间为24h。当反应结束后对其进行冷却,直到温度为室温为止,反应液减压浓缩,利用15mL的热水和15mL的EA进行萃取,次数为3次,合并有机相。在干燥过程中,选择利用无水Na2SO4完成,减压浓缩获得粗产物。利用比例为5:1的PE:EA打浆,通过过滤最终得到黄棕色固体40,含量为0.198g,收率达到72%。
结合最终成效来看,通过对这一工艺的应用,能获得较为良好的效果,在整个合成过程中,不需要增加额外设备,工艺改动幅度较小,可以通过简单的方式操作,能获得高质量的产品,可以为药品申报提供更大便利。从整体的角度分析,加强对药物中间体合成工艺的创新与优化,可以让中间体流失问题得到有效解决,也能减少原料的损耗,能够让工业批量生产需求得到满足。
4.结论
综合而言,在药物中间体合成期间,可以应用的技术工艺类型较多,包括缩合、定向硝化等,具体应用何种技术工艺还需要结合实际生产需求,有针对性地加以利用。随着药品中间体更新速度的不断加快,合成技术也要适当创新和优化,科学选择合成工艺路线,确保中间体生产效率在整体提升的同时,合成成本也能控制在合理范围内,让化工生产精细化的要求得到满足,为我国药物生产工艺技术的深入发展奠定良好基础。
