虎杖解毒颗粒(无糖型)的质量标准研究Δ
2022-09-06白雨鑫童荣生李晋齐
白雨鑫,童荣生,李晋齐
(1.四川省骨科医院药学部,成都 610072; 2.四川省人民医院药学部,成都610072)
虎杖解毒颗粒(无糖型)为四川省人民医院的医院制剂(批准文号为川药制字Z20080173),已在临床应用30余年,清热、解毒疗效显著,广受患者好评[1-2]。根据临床实际需要,结合该制剂载药量的要求,拟研制成无糖颗粒。课题组前期开展了虎杖解毒颗粒(无糖型)成型工艺研究,获得了稳定可行的制备工艺[3]。为保证工艺变更后产品质量稳定可靠,本研究对虎杖解毒颗粒(无糖型)的质量标准进行了研究。根据《中华人民共和国药典:四部》(2020年版)通则[4],测定其粒径、水分、溶化性和装量差异,采用薄层色谱法定性鉴别虎杖解毒颗粒(无糖型)中的虎杖、板蓝根和川射干3味药材,采用高效液相色谱法定量测定颗粒中虎杖苷、大黄素的含量,采用加速稳定性实验对颗粒样品进行稳定性考察,以期为虎杖解毒颗粒(无糖型)的质量控制提供依据[5-7]。
1 材料
1.1 仪器
Waters e2695型高效液相色谱仪,Waters 2998PDA型检测器,Empower3色谱数据工作站(美国 Waters公司);BP211D型十万分之一电子分析天平(德国Sartorius公司);优普ULPHW型纯水机(成都超纯科技有限公司);KX-85-2A数显恒温磁力搅拌器(郑州南北仪器设备有限公司);双框冲压型标准药典筛(浙江绍兴市不锈钢筛厂,1号、3号和5号药典筛)。
1.2 药品与试剂
虎杖苷(批号:11575-200502)、L-精氨酸(C6H14N4O2)、大黄素(批号:110756-200110)、大黄素甲醚(批号:110578-200610)、射干苷(批号:111632-200501)、虎杖对照药材(批号:110980-200902)、板蓝根对照药材(批号:121177-201006)和川射干对照药材(批号:110264-200902)均购自中国食品药品检定研究院;薄层层析硅胶板(青岛海洋化工厂分厂);甲醇、乙腈为色谱纯(美国DIKMA公司);磷酸、乙酸乙酯、石油醚、甲酸乙酯、甲酸甲酯、正丁醇、冰醋酸、浓硫酸、三氯甲烷、丁酮和乙醚为分析纯,均购于成都市科龙化工试剂厂;试验用水超纯水。
2 方法与结果
2.1 性状评价
本品为棕黄色至棕色颗粒,颗粒大小适中,色泽均匀,气微香,味微甜,无吸潮、结块现象。
2.2 粒度检查
称取3批颗粒各50 g,按《中华人民共和国药典:四部》(2020年版)通则“物理检查法0982”(第二法双筛分法)下的方法进行测定。结果显示,3批颗粒中不合格颗粒均未超过颗粒总量的15%,符合药典规定。
2.3 溶化性检查
称取3批颗粒各10 g,加热水200 mL(70 ℃),用磁力搅拌器以固定转速搅拌,搅拌5 min,立即观察。结果显示,3批颗粒全部溶化且无异物,符合药典规定。
2.4 水分检查
称取3批颗粒各2 g,按《中华人民共和国药典:四部》(2020年版)通则“水分测定法0832”(第二法烘干法)进行测定。结果显示,3批颗粒的含水率均未超过6.0%,符合药典规定。
2.5 装量差异检查
从3批颗粒中各随机抽取10袋,去除包装,分别称定每袋内容物的重量,并与标示重量比较。结果显示,3批样品颗粒的装量差异<±5%,符合药典规定。
2.6 鉴别
参考《中华人民共和国药典:四部》(2020年版)及大量相关文献中资料,应用通则“0502薄层色谱法”对方中药材进行定性鉴别。结果显示,虎杖、川射干和板蓝根色谱斑点显色清晰,可作为制剂质量鉴别,但白芷、贯众和青蒿专属性不佳,未能纳入质量标准草案鉴别项。
2.6.1 虎杖的薄层鉴定:参照相关方法[8-11],将虎杖解毒颗粒(无糖型)研细后过三号筛,称取过筛粉末0.3 g,加入甲醇20 mL,超声(功率720 W,频率40 kHz)1 h后滤过,挥干滤液,向残渣中加入硫酸(2.5 mol/L)和氯仿各10 mL,100 ℃条件下加热回流1 h,放冷,转移液体至分液漏斗中,加入氯仿10 mL振摇,收集氯仿液,重复2次后合并氯仿液,水浴蒸干,用1 mL氯仿液溶解残渣,即得供试液。处方中去除虎杖药材,以原工艺制备阴性样品,称取阴性样品0.3 g、虎杖对照药材0.1 g,按供试液制备法制得阴性对照溶液、对照药材溶液。以甲醇为溶剂制备0.5 mg/mL的大黄素对照品溶液、大黄素甲醚对照品溶液。吸取上述5种溶液,各5 μL,在同一硅胶GF薄层板上点样(样品液点样2份),展开剂选定2种,分别为石油醚-甲酸乙酯-甲酸(V∶V∶V=15∶ 5∶ 1)的上层溶液和石油醚-甲酸甲酯-甲酸(V∶V∶V=15∶ 5∶ 1)的上层溶液[12]。跑板结束后取出晾干,置紫外灯(365 nm)下检视。结果显示,供试品在对照药材与大黄素、大黄素甲醚对照品的薄层色谱的相同位置上显示相同颜色的斑点,阴性对照品的相应位置没有斑点,见图1。

A.展开剂1;B.展开剂2;1.对照药材;2.阴性对照品;3.大黄素甲醚;4.大黄素;5—6.供试品A. developing solvent 1; B. developing solvent 2; 1. control reference; 2. negative control reference; 3. physcion; 4. emodin; 5-6. samples for test
2.6.2 板蓝根的薄层鉴别:参考相关方法[13-14],将虎杖解毒颗粒(无糖型)研细后过三号筛,称取过筛粉末0.5 g,加入稀乙醇20 mL,超声(功率720 W,频率40 kHz)1 h后滤过,挥干滤液,加稀乙醇1 mL溶解残渣,即得供试液。处方中去除板蓝根药材,以原工艺制备阴性样品,称取阴性样品0.5 g、板蓝根对照药材0.2 g,按供试液制备法制得阴性对照溶液、对照药材溶液。以稀乙醇为溶剂制备0.5 mg/mL的精氨酸对照品溶液。吸取上述4种溶液,各5 μL,在同一硅胶GF薄层板上点样(样品液点样2份),展开剂选定2种,分别为正丁醇-冰醋酸-水(V∶V∶V=19∶ 5∶ 5)和正丁醇-冰醋酸-水(V∶V∶V=22∶ 6∶ 3),跑板结束后取出晾干,喷以茚三酮显色试液,吹风加热至斑点清晰。结果显示,日光灯下,供试品在对照药材与精氨酸对照品的薄层色谱的相同位置上显示相同颜色的斑点,阴性对照品的相应位置没有斑点,见图2。
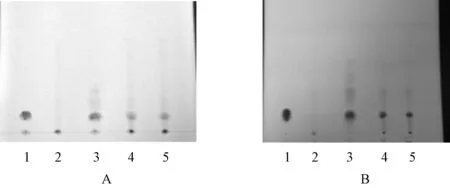
A.展开剂1;B.展开剂2;1.精氨酸;2.阴性对照品;3.对照药材;4—5.供试品A. developing solvent 1; B. developing solvent 2; 1. arginine; 2. negative control reference; 3. control reference; 4-5. samples for test
2.6.3 川射干的薄层鉴别:参考相关方法[15],将虎杖解毒颗粒(无糖型)研细后过三号筛,称取过筛粉末0.5 g,加甲醇10 mL,超声(功率720 W,频率40 kHz)0.5 h后滤过,挥干滤液,加1 mL甲醇溶解残渣,即得供试液。处方中去除川射干药材,以原工艺制备阴性样品,称取阴性样品0.5 g、川射干对照药材0.5 g,按供试液制备法制得阴性对照溶液、对照药材溶液。以甲醇为溶剂制备0.5 mg/mL的射干苷对照品溶液。吸取上述4种溶液,各5 μL,在同一硅胶GF薄层板上点样(样品液点样2份),展开剂选定2种,分别为三氯甲烷-丁酮-甲醇(V∶V∶V=3∶ 1∶ 1)和三氯甲烷-冰醋酸-甲醇(V∶V∶V=8∶ 0.2∶ 2),跑板结束后取出晾干,置紫外线灯(254 nm)下检视。结果显示,供试品在对照药材与射干苷对照品的薄层色谱的相同位置上显示斑点,阴性对照品的相应位置没有斑点,见图3。

A.展开剂1;B.展开剂2;1.对照药材;2.阴性对照品;3.射干苷;4—5.供试品A. developing solvent 1; B. developing solvent 2; 1. control reference; 2. negative control reference; 3. tectoridin; 4-5. samples for test
2.7 含量测定
2.7.1 虎杖苷含量测定:(1)色谱条件。色谱柱为Phenom Gemini C18色谱柱(250 mm×4.6 mm,5 μm);流动相为乙腈-水(V∶V=15∶ 85);检测波长为306 nm;流速为1.0 mL/min;柱温为30 ℃;进样量为10 μL。(2)溶液的制备。将虎杖解毒颗粒(无糖型)研细后过三号筛,精密称定过筛粉末(约0.5 g)入棕色容量瓶中,加甲醇40 mL,超声(功率720 W,频率40 kHz)1 h,放冷再精密定容,摇匀溶液,经0.45 μm有机系微孔滤膜滤过,续滤液即为供试品溶液[16]。处方中去除虎杖药材,以原工艺制备阴性样品,按供试品溶液制备方法制得阴性对照溶液。精密称取干燥后的虎杖苷对照品,加甲醇制得8.83 μg/mL的对照品溶液。(3)系统适用性实验。取虎杖苷对照品溶液、供试品溶液和阴性对照溶液,在上述色谱条件下进样。结果显示,虎杖苷色谱峰与其他组分分离良好,与其相邻色谱峰的分离度>1.5,阴性对照溶液在虎杖苷色谱峰位置上无干扰,虎杖苷保留时间为24.738 min,该法具有良好的专属性,见图4。(4)方法学考察。采用高效液相色谱法,精密吸取上述虎杖苷对照品溶液2、4、6、8、10、14、18和20 μL,进样测定。结果显示,虎杖苷在0.017 6 6~0.176 6 μg范围内线性关系良好(r=0.999 8)。连续进样6次,虎杖苷平均含量为1.071 5 mg/g,RSD为0.95%,仪器精密度良好。稳定性、重复性考察结果的RSD分别为1.22%、1.12%。加样回收率考察结果平均值为98.79%,RSD为1.97%。(5)样品测定。取3批虎杖解毒颗粒(无糖型)样品,每批取3份,每份0.5 g,制备供试品溶液,进样测定。计算得出不同批次虎杖苷含量范围为1.064 2~1.075 6 mg/g,平均含量为1.070 3 mg/g,RSD为0.54%。根据上述试验结果,考虑到药材的来源、制剂生产和贮藏等因素,暂定本品每袋含虎杖以虎杖苷计,不得少于8 mg/g。
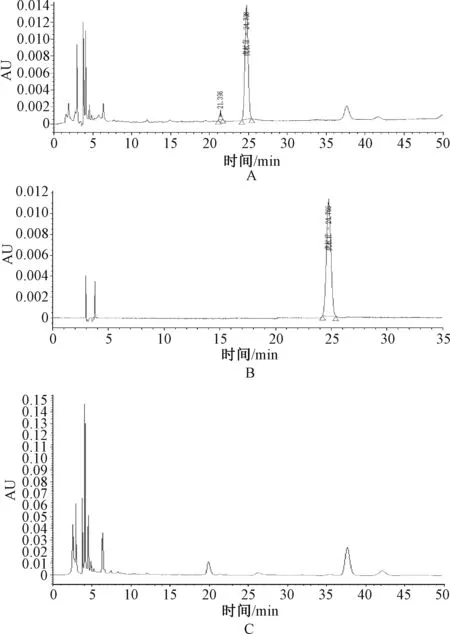
A.虎杖解毒颗粒(无糖型)供试品;B.虎杖苷对照品;C.虎杖苷阴性对照品A. sample of Huzhang antidote granules (sugar-free); B. reference substance of polydatin; C. negative control reference of polydatin
2.7.2 大黄素含量测定:(1)色谱条件。色谱柱为Phenomenex Gemini C18色谱柱(250 mm×4.6 mm,5 μm);流动相为甲醇-0.1%磷酸溶液(V∶V=72∶ 28);检测波长为254 nm;流速为1.0 mL/min;柱温为30 ℃;进样量为10 μL[17]。(2)溶液的制备。将虎杖解毒颗粒(无糖型)研细后过三号筛,精密称定过筛粉末(约0.5 g)入棕色容量瓶中,加40 mL甲醇,超声(功率720 W,频率40 kHz)1 h,放冷再精密定容,摇匀溶液,经0.45 μm有机系微孔滤膜滤过,精确量取9 mL续滤液,挥干滤液,向残渣中加入硫酸(2.5 mol/L)和氯仿各10 mL,100 ℃条件下加热回流1 h,放冷,转移液体至分液漏斗中,加入氯仿10 mL振摇,收集氯仿液,重复2次后合并氯仿液,水浴挥干,用甲醇溶解残渣并精密定容至10 mL容量瓶中,经0.45 μm微孔滤膜滤过,续滤液即为供试品溶液[18]。处方中去除虎杖药材,以原工艺制备阴性样品,参照供试品溶液制备方法制得阴性对照溶液。精密称取干燥后的大黄素对照品,加甲醇制得7.28 g/mL的对照品溶液。(3)系统适用性实验。取大黄素对照品溶液、供试品溶液及阴性对照溶液,以上述色谱条件进样。结果显示,大黄素色谱峰与其相邻色谱峰的分离度>1.5,大黄素保留时间为16.803 min,阴性对照溶液在该位置上无干扰,该法具备良好专属性,见图5。(4)方法学考察。利用高效液相色谱,精密吸取上述大黄素对照品溶液2、4、6、8、10、14、18和20 μL,进样测定。结果显示,大黄素在0.014 56~0.145 6 μg范围内线性关系良好(r=0.999 9)。连续进样6次,大黄素平均含量为0.675 9 mg/g,RSD为0.68%,仪器精密度良好。稳定性、重复性考察结果的RSD分别为1.7%、1.67%。加样回收率考察结果平均值为101.53%,RSD为1.63%。(5)样品测定。取虎杖解毒颗粒(无糖型)样品3批,每批3份,每份0.5 g,制备供试品溶液,进样测定。结果显示,不同批次的大黄素含量范围为0.682 4~0.701 2 mg/g,平均含量为0.694 2 mg/g,RSD为1.48%。根据上述试验结果,考虑到药材的来源、制剂生产和贮藏等因素,暂定本品每袋含虎杖以大黄素计,不得少于5 mg/g。
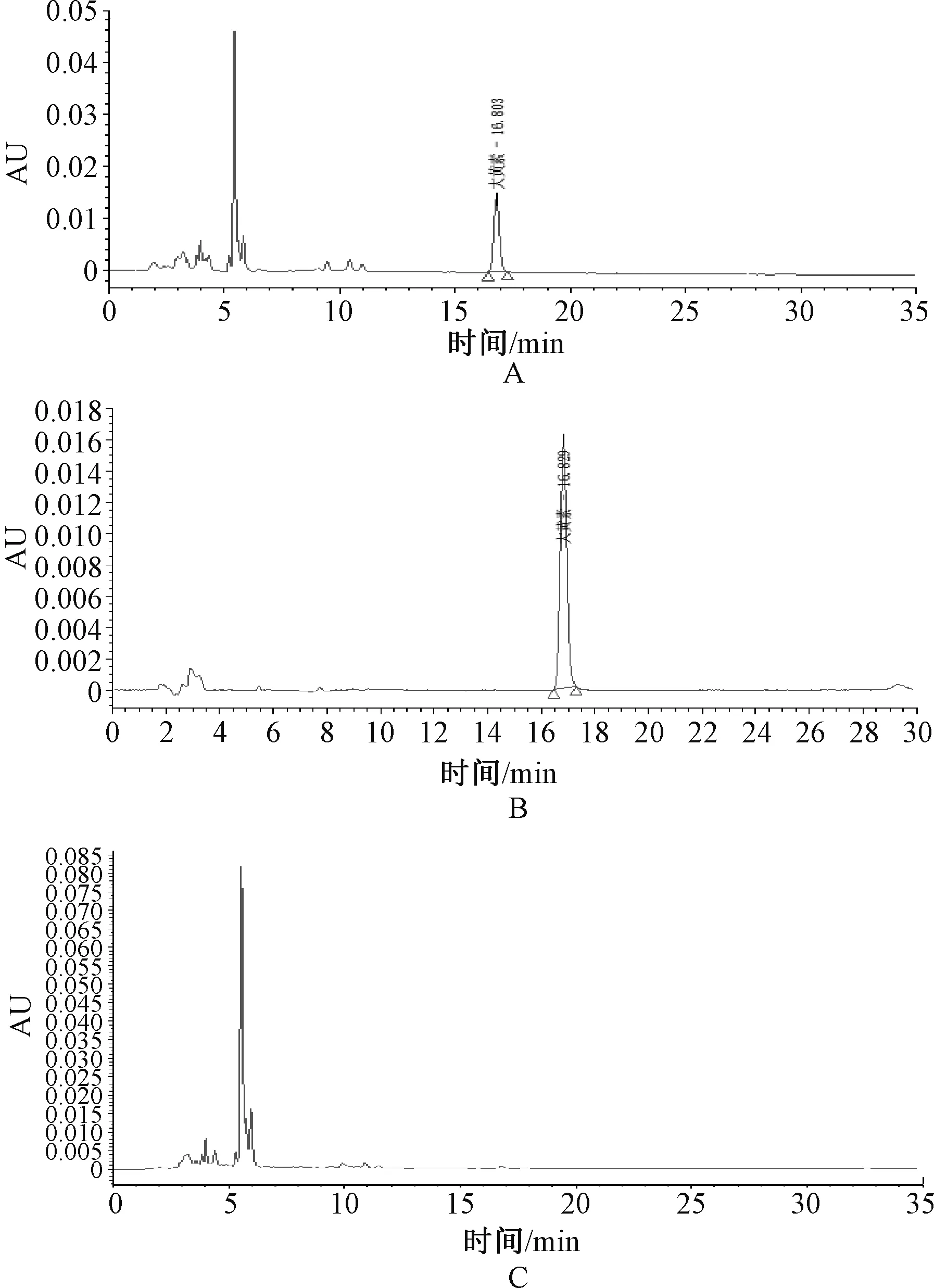
A.虎杖解毒颗粒(无糖型)供试品;B.大黄素对照品;C.大黄素阴性对照品A. sample of Huzhang antidote granules (sugar-free); B. reference substance of emodin; C. negative control reference of emodin
2.8 虎杖解毒颗粒(无糖型)的加速稳定性实验
为考察颗粒质量稳定性,将3批样品(市售包装),置温度为40 ℃±2 ℃,相对湿度为75%±5%的恒温恒湿培养箱中,于第1个月、2个月和3个月取样,进行含量测定和薄层鉴别,并考察记录颗粒性状、粒度、水分、溶化性和装量差异情况,结果见表1。与0个月考察结果进行对比,在实验期内,3批制剂样品的各项检查指标均符合规定,在加速稳定性实验条件下质量基本稳定[19]。
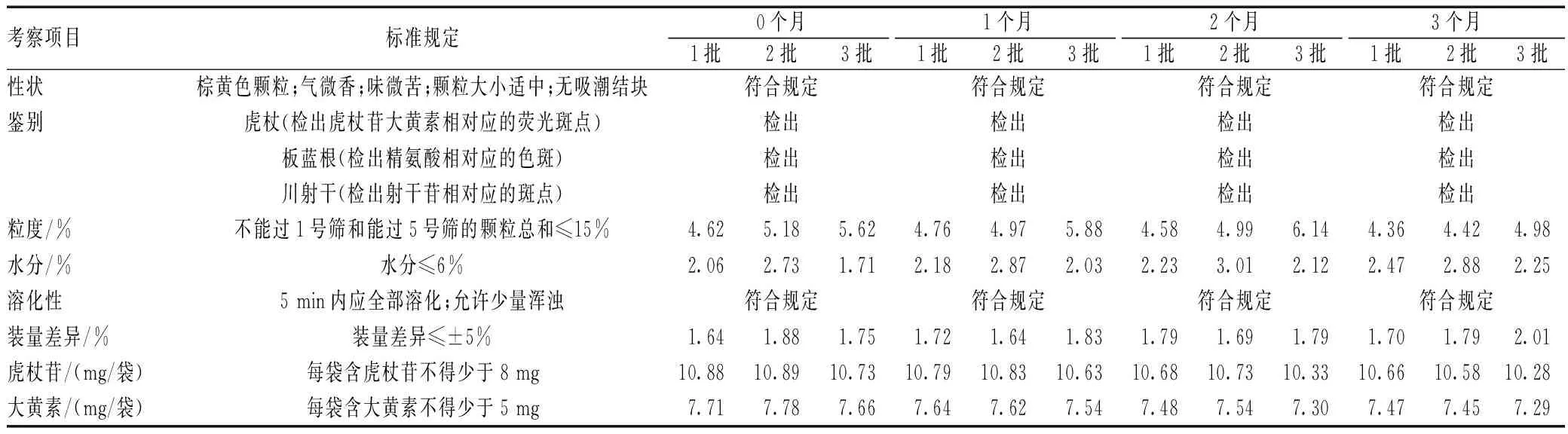
表1 加速稳定性实验结果Tab 1 Results of accelerated stability
3 讨论
3.1 颗粒剂质量检查
参照《中华人民共和国药典:四部》(2020年版),检查了3批颗粒样品的粒径、溶化性、水分和装量的差异,结果表明,颗粒质量符合规定。
3.2 薄层色谱法鉴别
为了控制制剂的质量,保证疗效,对处方中的药材进行薄层鉴别。为确保鉴别结果科学可靠,本研究对方中虎杖、川射干和板蓝根均选择2种不同的展开剂进行分离。结果显示,不同展开剂条件下分离效果均理想,斑点清晰,阴性无干扰;并且3批颗粒样品结果一致,表明鉴别方法稳定可行,重现性高,可作为该颗粒的质量控制方法。
3.3 含量测定
本研究中,虎杖苷和大黄素的含量测定直接参照《中华人民共和国药典:四部》(2020年版)虎杖的含量测定方法。实验发现,当虎杖苷采用药典流动相条件(乙腈-水,V∶V=23∶ 77)时,杂质干扰很大,虎杖苷不能与相邻的色谱峰完全分离。根据经验和对实验条件的不断探索,以乙腈-水(V∶V=15∶ 85)为流动相,以去除其他成分的干扰。
综上所述,本研究对虎杖解毒颗粒(无糖型)的质量进行了考察,建立了虎杖、板蓝根和川射干共3味中药的薄层色谱鉴别法,以及以虎杖苷、大黄素为指标成分的高效液相色谱含量测定方法。加速稳定性实验结果证明制剂具有较好的稳定性,工业生产后可长期保质存放,有效期的最终确立可待继续考察。本研究为虎杖解毒颗粒(无糖型)的质量研究提供了实验依据,建立的方法准确可靠、专属性强、重复性好,可用于虎杖解毒颗粒(无糖型)的质量控制。
