202例急性淋巴细胞白血病儿童细胞遗传学特点
2022-09-05尹萌萌刘爱国张艾王雅琴胡群
尹萌萌 刘爱国 张艾 王雅琴 胡群
急性淋巴细胞性白血病(acute lymphoblastic leukemia,ALL)为儿童时期常见的血液肿瘤疾病,在过去的十年中,分子生物学及细胞遗传学的大力发展使人们发现更多有关白血病的染色体核型及基因,增强了人们对白血病的生物学认识,包括发病机制、诊断及预后等,促进了临床对患儿的精确个体化治疗[1]。以往研究证实ALL儿童携带的染色体核型及融合基因,可作为白血病诊断和预测疾病进展的生物标志物[2],但其在病人群体的分布特点,以及与患儿临床特征的相关性,还有待进一步研究。因此,本次研究对202例ALL儿童的细胞遗传学特点进行研究,分析其与临床特征的相关性。
对象与方法
1.研究对象
本科室2009~2015年诊断的202例ALL儿童,随访至2018年底,中位随访时间为5.9年。将本组ALL患儿分成3个年龄组:① 年龄≤1岁,②1岁<年龄≤6岁,③年龄>6岁。收集患儿细胞遗传学结果及临床资料,包括性别、年龄、临床危险度及预后等。
2.ALL诊断及临床危险度分型标准
ALL诊断标准:骨髓涂片中,淋巴系细胞增殖异常,原始+幼稚淋巴细胞≥30%骨髓有核细胞,即可诊断ALL。
临床危险度分型标准:(1)低危ALL(low risk-ALL,LR-ALL):不具有中危及高危因素。(2)中危ALL(intermediate risk-ALL,IR-ALL):具有以下1项或多项者:①年龄≥10岁;②初诊时白细胞≥50×109/L;③中枢神经系统白血病;④T-ALL;⑤染色体数<45,或除t(12;21)、t(9;22)以外的异常染色体核型,或除t(4;11)以外的MLL基因重排。(3)高危ALL(high risk-ALL,HR-ALL):具有以下1项或多项者:①年龄<12个月;②初诊时白细胞≥100×109/L;③有t(9;22)染色体核型(BCR-ABL1融合基因)或t(4;11)染色体核型(MLL-F4融合基因);④诱导缓解治疗效果不佳。
3.检测方法
(1)染色体核型:取骨髓细胞,采取R显带法后,显微镜下观察中期分裂相细胞染色体核型,核型定义参照《人类细胞遗传学国际命名法体制(ISCN)1995》。
(2)免疫分型:选择免疫荧光法和流式细胞术,采用的单克隆抗体包括:T-ALL系:CD1、CD2、CD3、CD4、CD5、CD7、CD8及TdT等。B-ALL系:CD10、CD19、CD20、CD23、及SmIg等。髓系:CD11b、CD13、CD14、CD15、CD33、CD117及cMPO等。
(3)融合基因:取患儿骨髓细胞,选择荧光原位杂交技术(FISH)或逆转录-聚合酶链反应(RT-PCR)技术检测融合基因。
4.治疗方案及预后分组
本组患儿均按照中华儿科杂志儿童急性淋巴细胞性白血病诊疗建议(第三次修订草案)治疗[3],异常染色体包括:超二倍体:染色体数目>46;亚二倍体:染色体数目<46。假二倍体:染色体数目为46条,存在结构异常者。
根据文献报告的染色体与预后关系将患儿进行分组[4,5]:
①预后良好组:超二倍体,t(12;21)及正常核型;
②预后中等组:t(1;19),假二倍体及其它简单结构异常核型;
③预后差组:t(9;22),t(4;11)及亚二倍体。
5.统计学方法:
采用SPSS19.0统计软件分析数据,分类资料以百分比表示,并采用卡方检验进行组内比较。Kaplan-Meier法评估患儿生存状态及绘制生存曲线,各组生存率采用Log-rank检验比较,P<0.05为差异有统计学意义。
结果
1.基本情况
202例ALL患儿中,男性 136例,女性 66例,男女比例为2.1:1。患儿年龄0.6~16岁,中位年龄为 5.4岁。
2.ALL儿童细胞遗传学特点
本组患儿中,正常核型占67.8%(137/202),异常核型占32.2%(65/202),其中单纯数目异常者占21.5%(14/65),单纯结构异常者占35.4%(23/65),数目及结构均异常者占43.1%(28/65)。
本组患儿中,超二倍体占46.2%(30/65),假二倍体占38.5%(25/65),亚二倍体占15.3%(10/65)。异常结构核型中,t(9;22)占9.8%(5/51),t(1;19)占9.8%(5/51),t(4;11)占3.9%(2/51),t(12;21)占2%(1/51),t(6;19)占3.9%(2/51),t(3;17)占3.9%(2/51),t(12;22)占2%(1/51),t(6;12)占3.9%(2/51)及其它改变核型占60.8%(31/51),见图1。
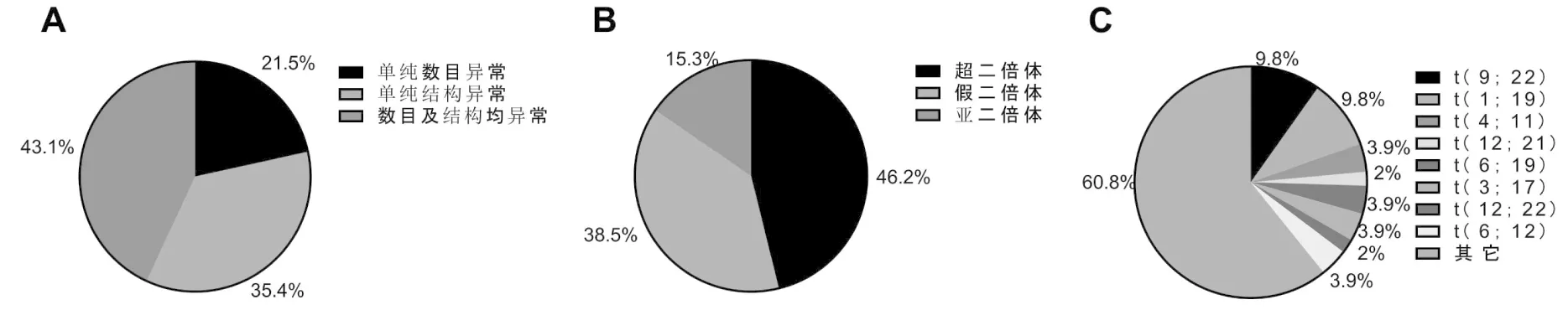
图1 不同类型染色体核型构成比例
融合基因阳性占20.8%(42/202)。42例融合基因中,TEL/AML1占42.9%(18/42),BCR/ABL1占23.8%,(10/42),E2A/PBX1占23.8%(10/42),SIL/TAL1占9.5%(4/42),见图2。
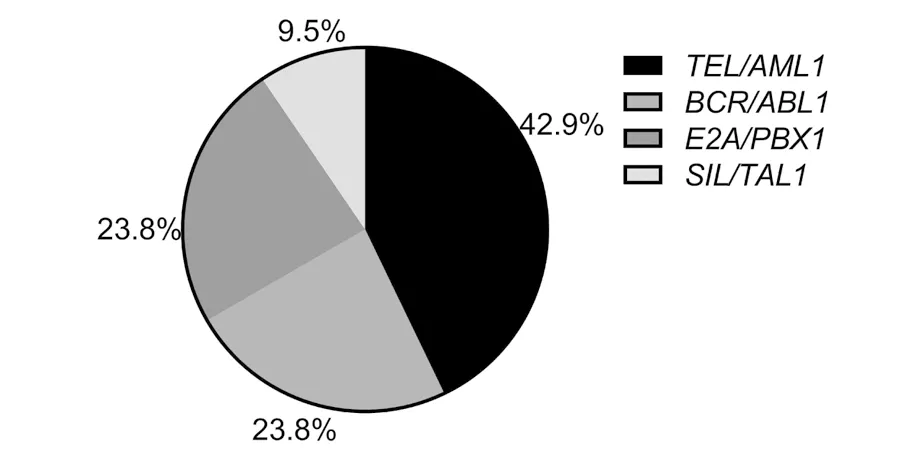
图2 ALL儿童融合基因构成比例
3.各年龄组ALL儿童细胞遗传学特点
本组患儿中,年龄≤1岁仅1例,1岁<年龄≤6岁共127例,年龄>6岁共74例。三组患儿的细胞遗传学资料比较后发现,1岁<年龄≤6岁组患儿的异常染色体(37%vs23%,P=0.039)及超二倍体比例(18.9%vs8.1%,P=0.038)高于年龄>6岁组患儿,但BCR/ABL1比例较低(2.4%vs9.5%,P=0.026),见表1。
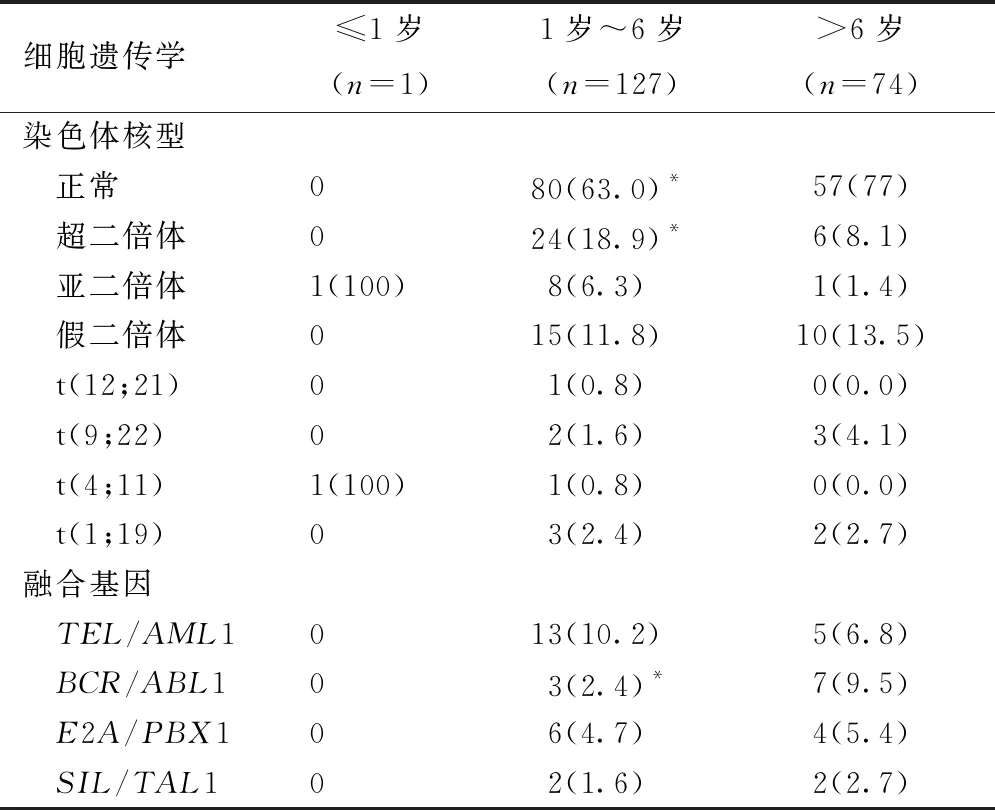
表1 不同年龄组ALL儿童细胞遗传学资料,n(%)
4.不同性别ALL儿童细胞遗传学特点
不同性别患儿之间的异常核型发生率无明显差别(29.4%vs37.9%,P=0.227),不同类型染色体及融合基因间分布无明显差别(均P>0.05),见表2。
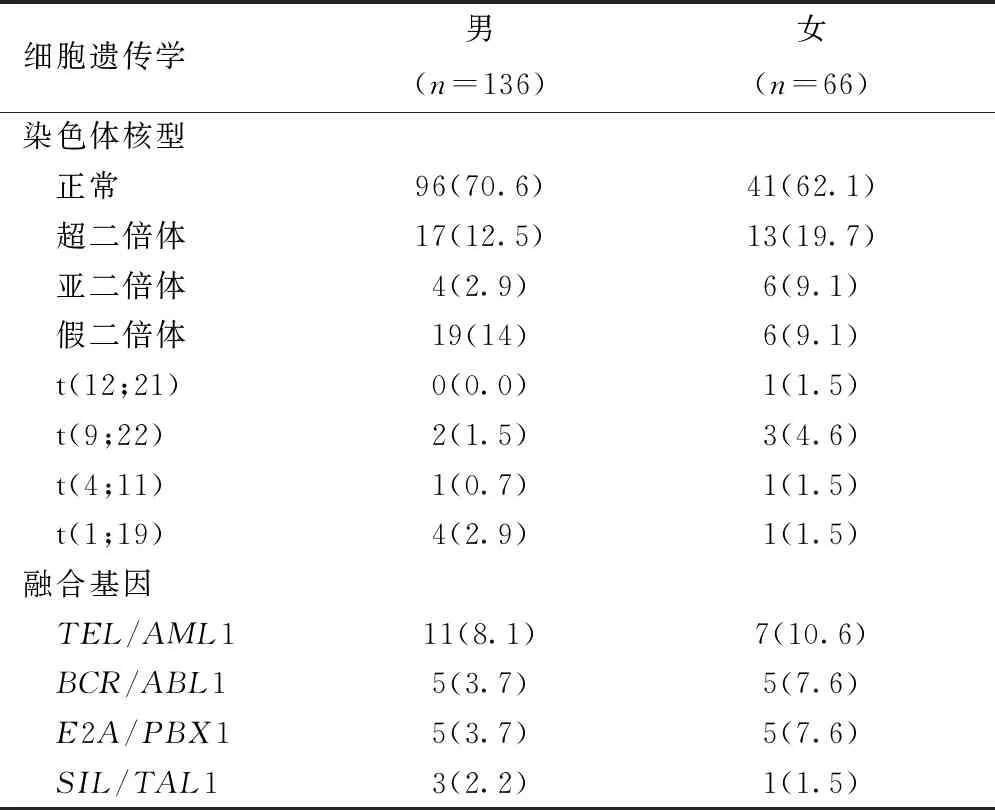
表2 不同性别ALL儿童细胞遗传学资料,n(%)
5.不同临床危险度ALL儿童染色体核型特点:
本组患儿中,LR 占25.7%(52/202),IR 占32.2%(65/202),HR占39.6%(80/202),未知占2.5%(5/202)。不同临床危险度患儿的细胞遗传学资料见表3,HR组中异常核型(42.5%vs40%vs9.6%,P<0.001)、亚二倍体(8.6%vs4.6%vs0%,P<0.001)及假二倍体比例明显高于另外两组患儿(22.6%vs9.2%vs1.9%,P<0.001),IR组中超二倍体比例明显高于另外两组患儿(26.2%vs11.3%vs7.7%,P<0.001)。
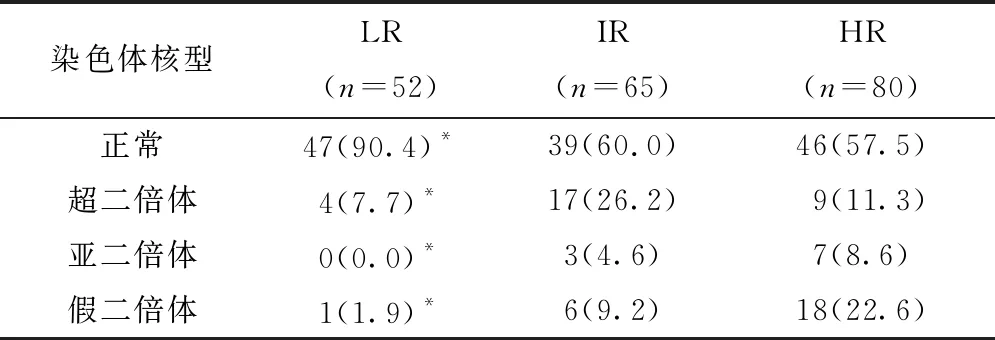
表3 不同临床危险度ALL儿童细胞遗传学资料,n(%)
6.融合基因与相应染色体核型比较
本组18例TEL/AML1阳性患儿中,超二倍体5例,亚二倍体1例,而t(12;21)仅有1例(5.6,1/18)。
10例BCR/ABL1阳性患儿中,检测出t(9;22)占50%(5/10)。携带t(9;22) 的患儿多伴有其它异常染色体结构,如-7/del(7p),9p异常,del(11p),del(9),dup(1q)和亚二倍体。
10例E2A/PBX1阳性患儿中,测出t(1;19)占50%(5/10),假二倍体3例,超二倍体2例,亚二倍体1例。
本组患儿未测出MLL基因,但测出2例患儿t(4;11)阳性。同时,4例SIL/TAL1患儿中,未发现6q杂合性缺失、del(1q32)等异常结构染色体核型。
42例融合基因阳性患儿中,26.2%(11/42)患儿携带对应的染色体核型。
7.临床预后分析
截止随访时间,存活166例(82.2%,166/202),死亡22例(10.9%,22/202),失访14例(6.9%,14/202)。统计不同染色体预后组的生存时间制作生存曲线,见图3。对本组患儿从ALL诊断时间(2009—2015年)开始随访,随访至2018.12.31,中位随访时间为5.9年。使用Log-rank检验比较三组患儿生存时间,具有明显统计学差异(P<0.001)。
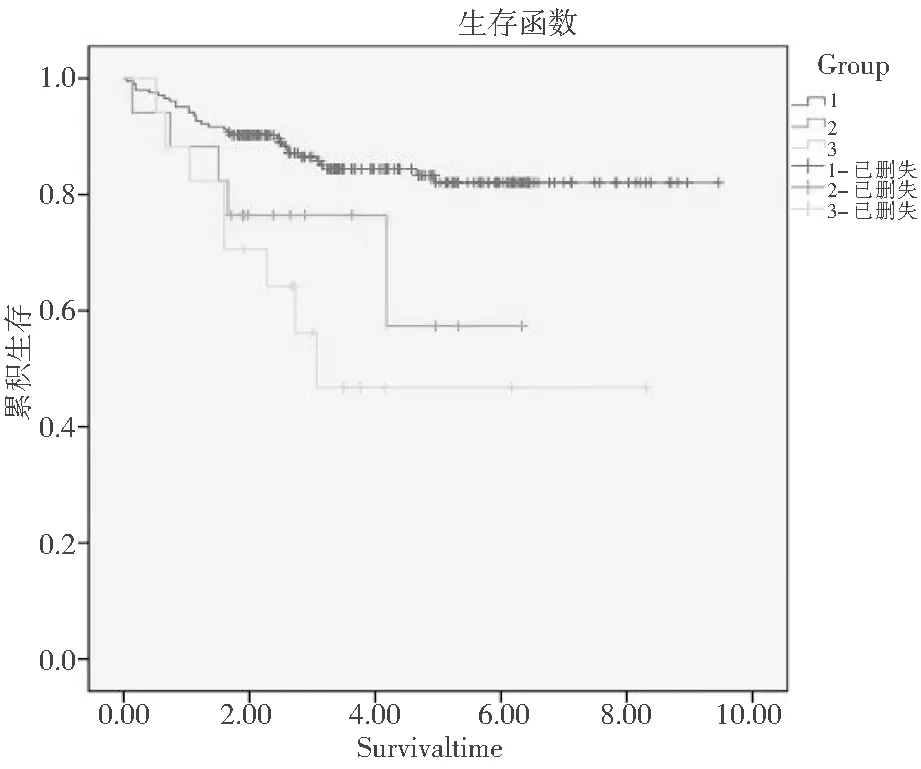
图3 三组不同染色体核型ALL患儿生存曲线比较
讨论
随着对ALL生物异质性认识的提高和生物检测技术的发展,ALL儿童的细胞遗传学特点得到了越来越多的关注。其中,染色体及基因与疾病进展、治疗及预后的相关性,已得到国际认可。文献中报道,ALL儿童的细胞遗传学改变发生率约为30%~50%[6],本组ALL儿童的染色体异常发生率为32.2%,基因突变率为20.8%,低于以往研究报道,可能是由于本组患儿诊断时间跨度较大,早期技术不成熟,而导致假阴性偏高。
染色体核型异常可表现在数目和/或结构改变。本组患儿中,数目及结构均异常者最多见(43.1%)。在异常遗传学结果中,代表预后良好的超二倍体(46.2%)及TEL/AML1(42.9%)最多见,证明ALL儿童总体预后较好。
1.不同融合基因检测及临床特点
BCR-ABL1重排(Ph+)对应的染色体易位为t(9;22),40%~85%的t(9;22) 易位患儿同时伴有其它结构异常[7],但同时存在的异常结构对预后的具体影响还尚无定论,有文献认为同时伴有-7/del(7p)或9p异常的预后更差,而伴随超二倍体者可改善预后[8]。本组中,5例患儿伴有t(9;22)易位,合并的异常核型分别为del(11p),dup(1q),del(9),-7/del(7p)和亚二倍体,与上述文献报道有差异。如今,酪氨酸激酶小分子抑制剂的靶向治疗可显著改善此类患儿预后[9]。本组病例中,BCR/ABL1基因阳性的患儿中仅一半患儿检测出t(9;22),可能是由于未观察到分裂中期骨髓细胞,或是检测技术不成熟导致[10]。
t(1;19)易位合并E2A-PBXl融合是儿童ALL中常见的染色体易位之一,伴t(1;19)异常的患儿,初诊时多有肿瘤浸润及高白细胞血症,预后欠佳,但近几十年来,早期高强度的化疗已改善t(1;19)患儿的预后[11]。本组患儿中,10例E2A/PBX1患儿仅测出5例t(1;19),仍低于基因检测阳性率。t(12;21)易位导致ETV6-RUNX1(TEL-AML1)基因融合,此类患儿预后较好[1]。与TEL基因有关的染色体易位包括t(9;12),t(12;22)和t(12;21),最常见的是t(12;21),发生于25%的B-ALL儿童中,但由于此易位位置隐蔽,常规染色体检查阳性率<1%,因此需结合FISH技术提高阳性检测率[10],本组仅检测出1例t(12;21)。
MLL基因中11q23重排各亚型因染色体易位不同具有明显的临床异质性,但此类患儿预后均欠佳,造血干细胞移植也无法显著改善其预后[12]。MLL基因中最常见的染色体改变为t(4;11)(q21;q23),此易位常伴有其它核型异常,特别是i(7)(q10)和+6,但并不进一步影响患儿的预后。11q23易位常见的断裂位点包括4q21,9p22,lp32,和19p13等[13]。本组患儿中,仅有1例婴儿,携带的染色体为t(4;11),但与其它基因阳性的患儿相反,未检测到MLL基因。可能在有关t(4;11)易位检测中,染色体改变比基因突变更易发现。
有文献表明,部分T-ALL儿童可携带SIL-TAL1基因,同时合并染色体6q杂合性缺失及del(1q32),此类患儿易发生高乳酸血症,但该融合基因对ALL的预后影响还未确定[14]。本次研究中,T-ALL患儿共20例,其中SIL-TAL1基因阳性4例,未发现6q杂合性缺失,del(1q32)等异常结构染色体核型,可能是受T-ALL病例数偏少影响。
在本组ALL儿童细胞遗传学检测中,42例阳性融合基因仅发现11例对应的染色体核型,一方面由于检测技术,一方面由于染色体核型改变的隐蔽性,但也存在2例t(4;11)易位患儿未发现MLL基因,说明在临床中,不仅存在融合基因检测阳性率高于染色体分析的现象,也说明ALL诊断准确性的提高,需要结合多种细胞遗传学方法综合判断。
2.不同临床特征ALL患儿细胞遗传学
了解了本组ALL儿童总体细胞遗传学改变特点后,根据患儿的不同临床特征分组,进一步具体分析不同组患儿中的细胞遗传学改变及分布特点。在临床中,可观察到ALL儿童的预后与年龄具有明显的相关性,如在治疗效果中,学龄前期及学龄期患儿优于婴儿期及青春期患儿。除了个人体质、药物耐受性及医嘱依从性的差异外,染色体及基因在不同年龄段患儿中是否也存在分布差别,需要我们比较分析。通过比较后发现,1岁<年龄≤6岁组患儿的异常染色体及超二倍体的分布比例高于年龄>6岁组患儿,而BCR/ABL1分布比例较低。本次分析结果不仅帮助临床更好了解细胞遗传学在不同年龄患儿中的分布差异,也进一步解释了学龄前期及学龄期患儿预后较好的原因。在不同性别患儿中,异常染色体及不同类型细胞遗传学结果分布均无差别,说明在ALL儿童中,男性与女性患儿的预后无明显差异,性别不能作为预后预测指标,同时在治疗方面也不需区别化疗。
ALL儿童的临床分型不仅取决于个人临床特征,也取决于某些特定的染色体及基因。但携带某些特定染色体及基因的患儿,大多同时合并其它异常染色体核型,因此我们分析了不同临床危险度患儿的染色体核型特点,经过比较后发现,HR组中异常核型、亚二倍体及假二倍体比例明显高于另外两组患儿,说明HR组患儿多同时合并预后不良的核型,但IR组中超二倍体比例高于另外两组患儿,一方面是由于HR组患儿中,预后良好的染色体核型偏少,另一方面是由于LR组患儿中,正常核型占有大部分。
本研究不仅发现染色体及基因在不同临床特征ALL儿童中的分布特点,也进一步证实了染色体与ALL预后的相关性,不仅强调了细胞遗传学在临床中的作用,也为我们在判断ALL儿童预后时,提供了指导性意见。
