Oxygen-deficient SnO2 nanoparticles with ultrathin carbon shell for efficient electrocatalytic N2 reduction
2022-08-30GungkiLiHeseongJngZijinLiJiWngXuqingJiMinGyuKimXienLiuJephilCho
Gungki Li,Heseong Jng,Zijin Li,Ji Wng,Xuqing Ji,c,,Min Gyu Kim,Xien Liu,,Jephil Cho
a State Key Laboratory Base of Eco-chemical Engineering,College of Chemical Engineering,College of Chemistry and Molecular Engineering,Qingdao University of Science and Technology,Qingdao,266042,China
b Department of Energy Engineering,School of Energy and Chemical Engineering,Ulsan National Institute of Science and Technology (UNIST),Ulsan,689-798,South Korea
c College of Materials Science and Engineering,Qingdao University,Qingdao,266071,China
d Beamline Research Division,Pohang Accelerator Laboratory (PAL),Pohang,790-784,South Korea
Abstract For high-efficiency NH3 synthesis via ambient-condition electrohydrogenation of inert N2,it is pivotal to ingeniously design an active electrocatalyst with multiple features of abundant surfacial deficiency,good conductivity and large surface area.Here,oxygen-deficient SnO2 nanoparticles encapsulated by ultrathin carbon layer(d-SnO2@C)are developed by hydrothermal deposition coupled with annealing process,as promising catalysts for ambient electrocatalytic N2 reduction.d-SnO2@C exhibits high activity and excellent selectivity for electrocatalytic conversion of N2 to NH3 in acidic electrolytes,with Faradic efficiency as high as 12.7% at -0.15 V versus the reversible hydrogen electrode(RHE) and large NH3 yield rate of 16.68 μg h-1at -0.25 V vs.RHE in 0.1 mol L-1 HCl.Benefiting from the structural superiority of enhanced charge transfer efficiency and optimized surface states,d-SnO2@C also achieves excellent long-term stability.
Keywords: N2 reduction reaction;NH3 synthesis;SnO2;Electrocatalysts;Ambient conditions
1.Introduction
Ammonia (NH3) is a vital building block to produce a series of important chemicals including fertilizer,medicine,and hydrogen storage molecules [1,2].NH3synthesis before the 1900s was mainly based on biological pathway where the nitrogenase is susceptible to inactivity,hampering industrial implementation [3].Artifical NH3production via atmospheric nitrogen reduction was invented in the 20th century [4],offering appealing routes but dominated by Haber-Bosch(HB)process with inevitable high temperatures (200-500°C) and pressures (150-300 atm) [5-7].The harsh conditions render HB approach is highly energy-intensive,accounting for 2%of world annual energy-consumption [8].And that indispensible steam-reforming of natural gas releases tremendous greenhouse gases(the global average is 2.86 tons of CO2per ton of NH3and 1.6 tons of CO2per ton of NH3in the most efficient plants) [9].Such severe climate impact and striking energy requirement accelerate the emergence of sustainable N2-fixation strategies to cope with ever-increasing NH3demands[10-12].
Electrochemical N2reduction has become one of the most competitive technologies due to its electro-driven operation relying on renewable sources at moderate temperature and atmospheric pressure[13].However,this electrochemical route is plagued by high enery barrier of N≡N bond breakage whose binding energy reaches up to 941 kJ mol-1[14].Designing efficient catalysts with robust activation ability to facilitate N≡N bond fracture while inhibiting undesired hydrogen evolution reaction (HER) is extremely essencial.Precious metalbased electrocatalysts,Ru[15],Rh[16],Au[17],Ag[18],etc.,have been reported to effectively catalyze nitrogen reduction reaction(NRR).Hollow gold nanocages demonstrates high NH3Faradaic efficiency(FE)of 30.2%[19].Even so,high-cost and low-abundance restrict their scalable applications.
Enlightened by iron(Fe)and molybdenum(Mo)cofactor of nitrogenase,Fe/Mo relevant heterogeneous electrocatalysts were constructed [20-26].Fe@Fe3O4hybirds achieve a NH3yield rate of 1.3×10-10mol s-1cm-2with FE of 6.25%[27].MoS2naosheets are active affording a NH3yield rate of 8.08 × 10-11mol s-1cm-2and FE of 1.17% [28].Relative lower N2-fixation performance for MoS2is attributed to intrinsic high HER activity,suppressing NRR process.To this end,SnO2is believed as a promising NRR electrocatalyst with no HER electroactivity [29].Sun and coworkers synthesized SnO2nanocubes with NH3FE of 2.17% [30].Liu et al.constructed fluorine-doped SnO2mesoporous nanosheets with NH3FE of 8.6%[31].Despite these advances,more efforts are still needed to boost the NRR activity of SnO2for industrial applications.It is known that nanoparticles with the reduced size can expose more catalytical active sites and high-index facets for better electrocatalysis [32,33].Moreover,defects/vacancy in ultrasmall nanoparticles can optimize electronic structure to enhance intrinsic activity[34].Engineering oxygen vacancy(Vo)on TiO2significantly boosted the NRR activity,permitting FE as high as 9.8%[35].WO3with tailored surface Vo achieves NH3yield rate of 4.2 μg h-1and FE of 6.8%[36].Ingeniously merging these advantages into one material system in a simple way toward high-performance NRR catalysis is highly challenging.
Herein,oxygen-deficient nanoparticles encapsulated by ultrathin carbon layer (d-SnO2@C) are designed and developed via hydrothermal deposition with subsequent annealing process as NRR electrocatalysts at ambient condition.d-SnO2@C exhibits high activity and excellent selectivity for electrocatalytic conversion of N2to NH3in acidic electrolytes,with NH3FE as high as 12.7% at -0.15 V versus the reversible hydrogen electrode(RHE)and large NH3yield rate of 16.68 μg h-1at -0.25 V vs.RHE in 0.1 mol L-1HCl.Meanwhile,it also shows strong electrochemical stability.
2.Experimental
2.1.Chemicals and materials
Tin (IV) chloride (SnCl4),D (+)-Anhydrous glucose,salicylic acid (C7H6O3),sodium citrate dehydrate(C6H5Na3O7·2H2O),sodium hydroxide (NaOH),sodium hypochlorite solution (NaClO),hydrazine monohydrate(N2H4·H2O),hydrochloric acid (HCl) and ethanol (C2H5OH)were purchased from China National Pharmaceutical Group Corporation.Sodium nitroferricyanide dihydrate (C5FeN6-Na2O2·H2O) and ammonium chloride (NH4Cl) were bought from Aladdin.Nafion solution (5 wt%) was purchased from Sigma-Aldrich.Nafion 117 membranes and carbon cloth were obtained from Shanghai Hesen Electric Co.,Ltd.All the chemicals were used without further purification.
2.2.Synthesis of d-SnO2@C
In a typical synthesis,3.75 g of D (+)-anhydrous glucose was dispersed in 30 mL of deionized water.Then 3.75 mmol SnCl4was added into above solution.The obtained homogeneous solution was transferred into a stainless-steel autoclave(40 mL) kept at 180°C for 12 h.When autoclave cooled naturally,solid power was collected by centrifugation and carefully washed with deionized water and C2H5OH.Dried in vacuum at 60°C for 2 h,the collected products were annealed at 550°C for 2 h in Ar atmosphere.Finally,the products were ready for subsequent characterizations.
2.3.Materials characterization
The X-ray diffraction (XRD) patterns were conducted on XRD system (D/Max2000,Rigaku,Japan).Scanning electron microscopy (SEM,Hitachi High-Technology Co.,Ltd.,Japan).TEM and HRTEM measurements were performed on a Tecnai G20 transmission electron microscope at an accelerating voltage of 120 kV.X-ray photoelectron spectroscopy(XPS,Escalab 250 xi) was used to investigate the chemical valence and composition of the prepared material.X-ray absorption near-edge structure (XANES) and extended x-ray absorption fine structure (EXAFS) were conducted with a BL 10 C beam line at Pohang Light Source (Korea) operated at 3.0 G eV with a 200-mA ring current.
2.4.Electrochemical measurements
Typically,the catalytic ink was a mixture of the prepared catalysts (3 mg),5 wt% Nafion (10 μL),ultrapure water(0.15 mL) and ethanol (0.3 mL).Then,the ink (35 μL) was loaded onto a piece of carbon paper (1 × 1 cm2).The mass loading was calculated to be 0.2 mg cm-2.Electrochemical NRR measurements were performed in 0.1 mol L-1HCl using a typical H-type cell separated by a Nafion 117 membrane.The Nafion membrane was pretreated in H2O2(5%),H2SO4(0.5 mol L-1) and ultrapure water in sequence at 80°C.Electrochemical NRR experiments were performed using a platinum sheet as the counter electrode,Ag/AgCl (saturated KCl)as the reference electrode and a carbon cloth loaded with the catalyst as the working electrode.All the potentials were calibrated to RHE according to the following formula: E (vs.RHE)=E(V vs.Ag/AgCl)+0.197 V+0.059×pH.All the polarization curves are the steady-state ones after several cyclic voltammetry cycles.Prior to electrocatalysis,the electrolytes were purged with nitrogen (purity: 99.999%) for 30 min.
2.5.Determination of NH3
NH3in the electrolyte was detected by UV-vis spectrophotometry with indophenol blue method.Detailed procedure is as follows: 4 mL electrolyte after potentiostatic test was mixed with 4 mL of 1 mol L-1NaOH solution containing C6H5Na3O7·2H2O(5 wt%)and C7H6O3(5 wt%),followed by the addition of 2 mL of NaClO solution (0.05 mol L-1) and 0.4 mL of C5FeN6Na2O solution (1 wt%).After 2 h in dark under room temperature,the solution was measured by UV-vis spectrophotometer at a wavelength of 655 nm.The calibration curve(y=0.10466x-0.0004,R2=0.999)presents good linear relation of the absorbance value with NH3concentration.The NH3yield rate was calculated based on the equation as follows:
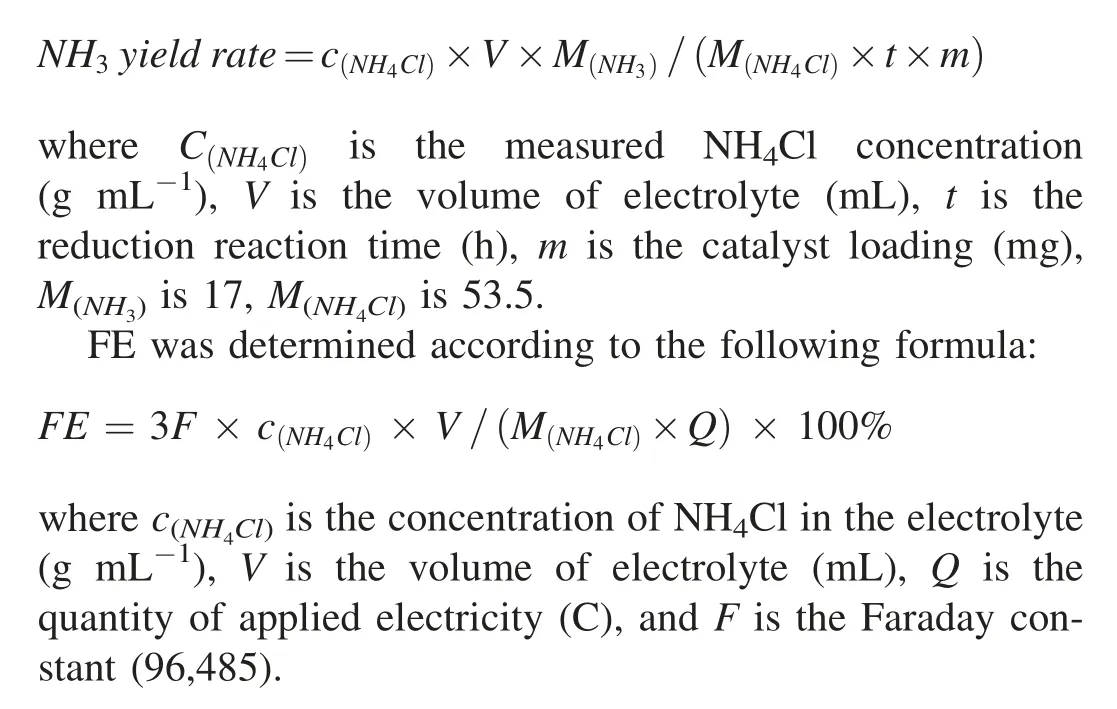
2.6.Determination of N2H4
N2H4was estimated by Watt and Chrisp method,and the color agent was obtained by mixing para-(dimenthylamino)benzaldehyde (5.99 g),concentrated HCl (30 mL) and C2H5OH (300 mL).Calibration curve was plotted as follows.N2H4standard solutions with the concentrations of 0.0,0.2,0.4,0.6,0.8 and 1.0 μg mL-1in 0.1 mol L-1HCl,were mixed with color reagent at the volume ratio of 5mL/5 mL.Staying in dark for 10 min,UV-vis absorbance of the mixed solution was measured at 455 nm.Good linear relationship between absorbance intensity and N2H4concentration was presented(y=1.17304x-0.00544,R2=0.999).
3.Results and discussion
d-SnO2@C was obtained via a facile two-step synthesis involving hydrothermal deposition of SnCl4and D (+)-anhydrous glucose coupled with annealing process.X-ray diffraction (XRD) pattern for as-synthesized d-SnO2@C is shown in Fig.1a.It is seen that d-SnO2@C presents diffraction peaks at 26.6°,33.8°,37.9°,51.7°,54.7°,61.8°,65.9°and 78.7°cor111responding to (100),(101),(200),(211),(220),(310),(301)and (312) crystal planes of tetragonal SnO2,respectively(JCPDS No.41-1445).The detailed chemical compositions and binding states of d-SnO2@C were further studied by X-ray photoelectron spectroscopy(XPS).All spectra were calibrated with the C 1s peak at 284.8 eV.XPS survey spectrum(Fig.1b)clearly evidences the presence of O,C,and Sn elements.Sn 3d can be deconvoluted into two peaks of 487.3 eV for Sn 3d5/2and 495.7 eV for Sn 3d3/2(Fig.1c) [37].In O 1s region,the peak at 530.6 eV is associated with lattice oxygen of d-SnO2@C [38].Associating peak at 531.2 eV can be assigned to oxygen vacancy,indicating the existence of defeciency[39].Binding energy of 532.2 eV is typical for the oxhydryl in adsorbed water on surface of d-SnO2@C (Fig.1d) [38].
d-SnO2@C was also investigated by scanning electron microscopy (SEM).The morphology of d-SnO2@C is spherical nanoparticle with diameter of ca.20-50 nm (Fig.2a and Fig.2b),which is benifical for more active catalytical sites.SEM image of precursor can be seen in Fig.S1.Clearly,its morphology is similar to d-SnO2@C.Transmission electron microscopy (TEM) images also confirm the spherical characteristic (Fig.2c and Fig.S2).High-resolution TEM(HRTEM)images taken from d-SnO2@C nanoparticle present well-resolved lattice fringes with interplanar distance of 0.335 nm (Fig.2d) cor111responding to (110) plane of SnO2.Ultrathin carbon layer of d-SnO2@C is observed in Fig.2e and Fig.S3,whose existence not only protects internal nanoparticles from aggregation but also enhances the conductivity[40].Energy-dispersive X-ray(EDX)spectrum for d-SnO2@C reveals that the ratio of O and Sn elements is 1.99:1,which is close to the expected stoichiometric ratio of 2 : 1 within the experimental error (Fig.S4).EDX spectroscopy elemental mapping images (Fig.2f-i) verify the uniform distribution of O,Sn and C elements in d-SnO2@C.All the characterizations prove the successful synthesis of d-SnO2@C.
We next measured the electrocatalytic NRR performances of d-SnO2@C in 0.1 mol L-1HCl under ambient condition using a two-compartment cell,which was separated by Nafion 117 membrane.All potentials are calibrated with respect to RHE.Prior to potentiostatic electrolysis,the potential range of d-SnO2@C for N2reduction was preliminarily evaluated via linear sweep voltammetry (LSV),and the electrolyte is 0.1 mol L-1HCl saturated with Ar and N2,respectively.Observed from LSV curves in Fig.3a,the current density of d-SnO2@C in N2-saturated electrolyte is relatively higher than that in Ar-saturated solution,reflecting that d-SnO2@C is active towards NRR.Therefore,we continue to eatimate its NRR performance by a continuous potentiostatic electrolysis of 1 h.The cor111responding curves are displayed in Fig.3b.The resultant-containing electrolytes were colored with indophenol blue method,and UV-Vis absorption spectra are presented in Fig.3c.Based on the calibration curve (Fig.S5),NH3yield rates and cor111responding FEs at different potentials are obtained and presented in Fig.3d.The maximum yield rate of NH3is 16.68 μg h-1at -0.25 V vs.RHE.NH3FE of d-SnO2@C catalyst could reach up to 12.7% at -0.15 V vs.RHE.The NRR performance of d-SnO2@C is excellent in compasion with some recent reported catalysts in acidic electrolyte (Table S1).On basis of Brunner-Emmet-Teller result(Fig.S6),d-SnO2@C has large specific surface area of 28.56 m2g-1,ensuring good active-site exposure.Possible by-product N2H4could not be detected(Fig.S7 and Fig.S8),disclosing high selectivity of d-SnO2@C for NH3synthesis.
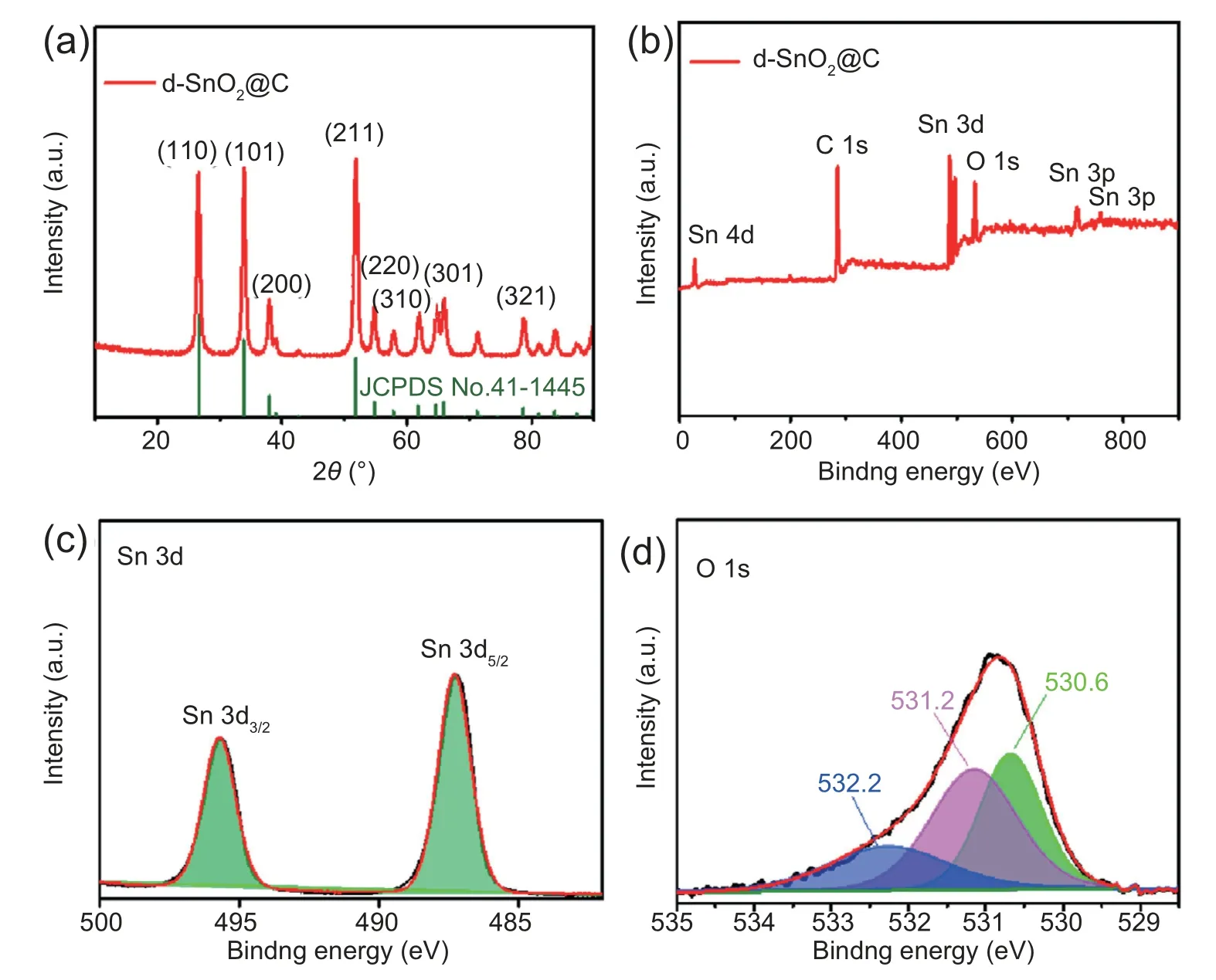
Fig.1.(a) XRD pattern of d-SnO2@C.(b) XPS survey spectrum of d-SnO2@C.XPS spectra of SnO2 in Sn 3d (c) and O 1s (d) regions.

Fig.2.(a,b) SEM images of d-SnO2@C with different magnifications.(c-e) HRTEM images of d-SnO2@C particles.(f-i) EDX elemental mapping images of d-SnO2@C.
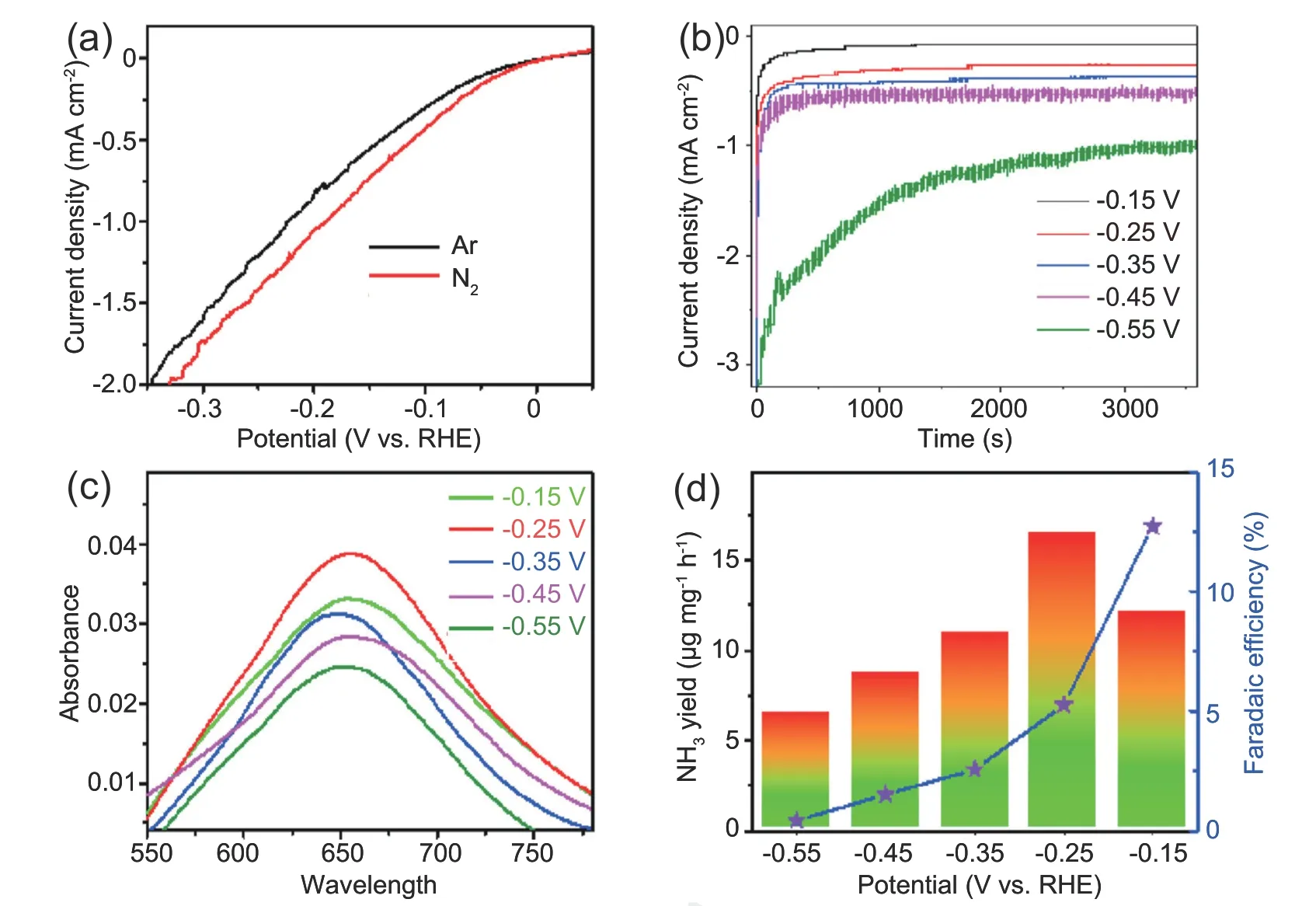
Fig.3.Electrochemical evaluation of the NRR performance of d-SnO2@C.(a)LSV curves in Ar/N2-saturated 0.1 mol L-1 HCl with a scan rate of 5 mV s-1.(b)Chronoamperometry curves at diverse potentials for 1 h in N2-saturated 0.1 mol L-1 HCl.(c)UV-vis adsorption spectra of the electrolytes stained with indophenol indicator after NRR electrolysis at different potentials for 1h.(d) NH3 yield rates and FEs at different potentials.
Comparative experiments were carried out to determine the origin of NH3from N2reduction.Observed from Fig.S9,pristine carbon paper(CC)substrate is inactive for N2-to-NH3process at standard conditions.Utilizing Ar atmosphere to achieve potentiostatic electrocatalysis of d-SnO2@C at-0.15 V vs.RHE,NH3cannot be obtained (Fig.S10).Obviously,N2is essential for NH3generation.To avoid NH3contaminat in N2source,N2-saturated electrolyte before potentiostatic electrolysis was stained with indophenol indicator.And that NH3signal was not observed in UV-vis adsorption spectrum (Fig.S11).All these results verify that NH3was synthesized by the NRR process.The durability of d-SnO2@C was tested by continuous catalytic reaction at-0.15 V vs.RHE for 24 h (Fig.4a).The catalytical current density is stable during the long-term testing.d-SnO2@C after 24 h stability test demonstrates almost no decline in ammonia yield rate and FE (Fig.S12).During successive four times repeated tests (Fig.4b),no significant fluctuation for NH3yield and FE of d-SnO2@C occurred,verifying that d-SnO2@C still retained high activity after long-term usage.
X-ray absorption near-edge structure (XANES) measurements were used to monitor structure changes of d-SnO2@C after NRR testing.XANES spectrum of Sn K-edge for d-SnO2@C before NRR process shows a main peak at 29,208 eV,which is similar to that of reported Sn4+species[41].After NRR test of d-SnO2@C,decreased peak intensity at 29,208 eV should be attributed to increased surface vacancies (Fig.4c) during NRR.Fig.4d shows the Fourier transforms (FT [k3w]) of Sn K-edge extended x-ray absorption fine structure (EXAFS) oscillations for d-SnO2@C before and after NRR process.The peaks at 1.6 Å can be attributed to Sn-O interaction [42].The lower intensity of Sn-O peak for d-SnO2@C after the NRR testing implies that the local structure around Sn atoms was further distorted during the NRR process [43],indicating the increase of oxygen vacancy.
Additionally,d-SnO2@C after 24 h stability test still demonstrates nanoparticle morphology (Fig.5 and Fig.S13),with XRD pattern of tetragonal SnO2(Fig.6a).XPS was also performed for d-SnO2@C after durability test(Fig.6b-d).For Sn 3d3/2and Sn 3d5/2,the binding energy is reduced from 487.3 eV to 495.7 eV-487.1 eVand 495.4 eV,showing that the oxidation state of Sn is reduced.By comparing XPS O1s spectra of d-SnO2@C before (Fig.1d) and after (Fig.6d) N2reduction,the increased peak intensity at 531.2 eV indicates higher Vo concentration in d-SnO2@C after NRR catalysis.The larger Vo concentration in post-NRR d-SnO2@C is attributed to the inevitable electrochemical reduction of the catalyst during long-term NRR catalysis at cathode potential[44].
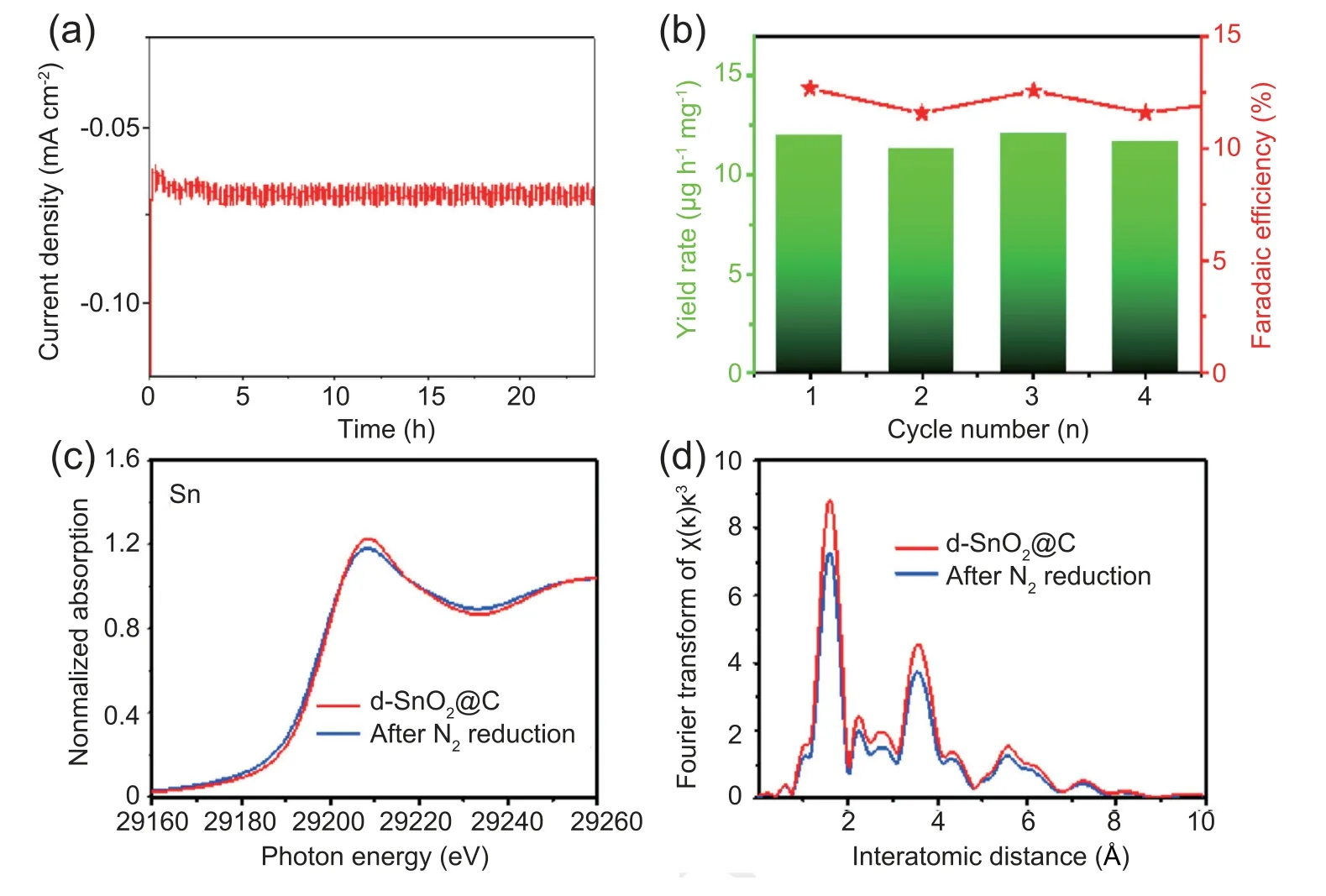
Fig.4.(a) Long-time chronoamperometry curve of d-SnO2@C at -0.15 V vs.RHE in N2-saturated HCl.(b) NH3 yield rate and FE of different test numbers at-0.15 V vs.RHE in N2-saturated 0.1 mol L-1 HCl after 1 h electrolysis.(c) XANES and (d) EXAFS of d-SnO2@C before and after stability test.
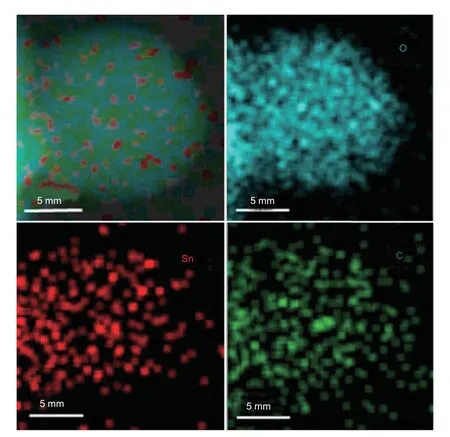
Fig.5.Scanning TEM and the energy-dispersive spectroscopic elemental mapping images of d-SnO2@C after stability measurements.
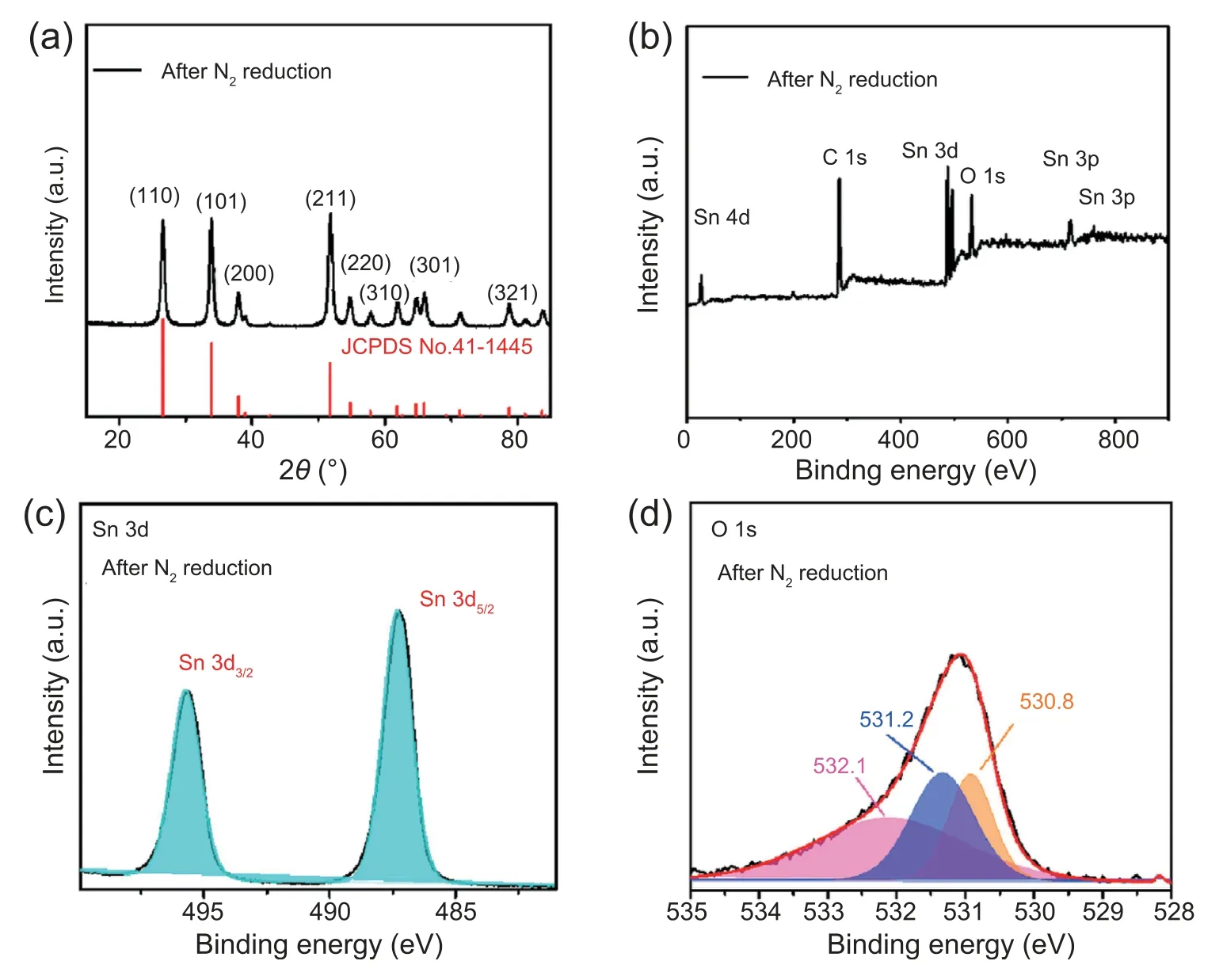
Fig.6.(a)XRD pattern of d-SnO2@C after long-term stability measurement.(b)XPS survey spectrum of the catalysts after durability test.High-resolution XPS spectra of Sn 3d (c) and O 1s (d) after durability test.
Pristine SnO2nanoparticles present poor NRR activity under acidic conditions(Fig.S14).To understand the key role of Vo for NRR,d-SnO2@C was heated in air to reduce the Vo content.As-obtained d-SnO2@C-Air and d-SnO2@C were charactrized by electron paramagnetic resonance (EPR),both exhibiting a pair of steep peaks (Fig.S15) with Vo signal at g=2.022 [35,36].d-SnO2@C-Air with reduced Vo content displays a significant reduction in NH3yield rate and FE(Fig.S16).The influence of Vo on NRR can be explained based on following reaction pathway: *N2→*NNH →*NNH2→*NHNH2→*NH2NH2→*NH2+NH3→*NH3+NH3→2NH3,where the proton and electron pair(H++e-)in each intermediate are omitted for clarity.*NNH formation step (*N2+H++e-→*NNH) is thermodynamically unfavorable [35].Vo in catalysts can stabilize the*NNH species considerably,optimizing the adsorption of N2.The increased N2chemisorption can suppress competing hydrogen evolution reaction to improve NH3FE.
4.Conclusions
In summary,d-SnO2@C catalyst with multiple structural advantages of abundant surfacial deficiency,good conductivity and large suface area is constructed by hydrothermal deposition followed by annealing process.As NRR electrocatalysts at ambient condition,d-SnO2@C exhibits high activity and excellent selectivity for electrocatalytic conversion of N2to NH3in acidic electrolytes,with NH3FE as high as 12.7% at-0.15 V vs.RHE and large NH3yield rate of 16.68 μg h-1at -0.25 V vs.RHE in 0.1 mol L-1HCl.This work provides a new perspective to design Sn-based catalysts for electrochemical reduction of N2to NH3under room temperature.
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by Taishan Scholar Program of Shandong Province,China(ts201712045);the Doctoral Found of QUST(0100229001)and 2019 Research Funds(1.190002.01)of Ulsan National Institute of Science and Technology(UNIST).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gee.2020.11.004.
杂志排行

Green Energy & Environment的其它文章
- Multivariate MOF for optimizing atmospheric water harvesting
- Lignin-based carbon fibers: Formation,modification and potential applications
- Charactering and optimizing cathode electrolytes interface for advanced rechargeable batteries: Promises and challenges
- Metal-organic frameworks-derived metal phosphides forelectrochemistry application
- Surface-mediated iron on porous cobalt oxide with high energy state for efficient water oxidation electrocatalysis
- Design and in-situ construct BiOI/Bi/TiO2 photocatalysts with metal-mediated heterostructures employing oxygen vacancies in TiO2 nanosheets