蒸馏后滴定法测定氯化钾中铵含量
2022-08-30刘伟朝程金莲
刘伟朝,程金莲
(1.青海和众网络科技有限公司,青海 西宁 810000;2.海西州盐化工产品质量检验检测中心,青海 格尔木 816000)
氯化钾被广泛应用于化工行业、农业、食品行业及电镀行业等[1]。当氯化钾作为原料生产其他产品时,对生产过程和产品质量都会产生一定的影响。氯碱生产过程中,原料氯化钾中带有一定量的铵,随原料被送去电解。电解过程中会生成三氯化氮,很可能因为某种原因导致爆炸事故发生,严重威胁着化工的安全生产[2]。作为原料生产食品添加剂氯化钾时,由于生产厂家不同,原料质量参差不齐,原料中杂质(铵)对生产过程和产品质量具有较大影响[3]。准确测定原料氯化钾中的杂质(铵)含量,可以指导企业调整生产工艺参数,提高产品质量。
目前,铵的测试方法有离子色谱法[4]、纳氏试剂分光光度法[5]、甲醛法[6]等,这些方法操作步骤较繁琐、耗时、使用的药品试剂较多,最重要的是纳氏试剂采用二氯化汞或碘化汞配制,此药品为剧毒物质,严重威胁着人的身体安全。文章中方法蒸馏后直接滴定,试验步骤简便,高效,且安全性高。
1 材料与方法
1.1 试剂与仪器
氢氧化钠(AR),硼酸(AR),盐酸(GR),甲基红(AR),亚甲基蓝(AR),碘化汞(AR),碘化钾(AR),均购于格尔木市博美化玻经销部;铵标准溶液(1 000 mg/L),购于坛墨质检科技股份有限公司。
氢氧化钠溶液(400 g/L):称取400 g氢氧化钠加水溶解后,冷却并稀释至1 000 mL,保存于塑料瓶中。硼酸溶液(20 g/L):称取20 g硼酸,加水溶解并稀释至1 000 mL。盐酸标准滴定储备溶液c(HCl)=0.5 mol/L,按照标准GB/T 601[7]中进行配制并标定。盐酸标准滴定溶液c(HCl)=0.05 mol/L,将盐酸标准滴定储备溶液稀释10倍。甲基红—亚甲基蓝混合指示液,2份甲基红乙醇溶液(1 g/L)与1份亚甲基蓝乙醇溶液(1 g/L)临用时混合。
紫外分光光度仪(美国Pekin Elmer),电子天平(德国赛多利斯公司),蒸馏装置(格尔木市博美化玻经销部)。
1.2 实验方法
1.2.1 蒸馏后滴定法
称取约10 g的试样(精确到0.000 1 g)于蒸馏烧瓶中,加约100 mL水,摇匀,加水至约400 mL,放入数粒爆沸珠。向接收器中准确加入20 mL硼酸溶液、4滴~5滴甲基红—亚甲基蓝混合指示液,加水至略高于接收器双连球管末端以保证封闭气体出口,将接收器连接在蒸馏装置的直形冷凝管下端。蒸馏装置的磨口连接处应涂硅脂密封。连接好蒸馏瓶,通过滴液漏斗加入40 mL氢氧化钠溶液,在溶液流尽时加入20 mL~30 mL水冲洗漏斗,剩3 mL~5 mL水时关闭活塞。打开冷却水,开始加热。蒸馏出至少150 mL馏出液后,用pH值试纸检查冷凝管出口的液滴,pH值不大于7结束蒸馏。关闭热源。将接收器中的溶液混匀,用盐酸标准滴定溶液滴定,直至指示液刚呈紫红色为终点。
1.2.2 纳氏试剂分光光度法
在6个50 mL比色管中,分别加入0.00 mL、1.00 mL、2.00 mL、3.00 mL、4.00 mL、5.00 mL的铵标准工作溶液(10 μg/mL),其所对应的铵含量为0.0 μg、10.0 μg、20.0 μg、30.0 μg、40.0 μg和50.0 μg,加水至标线。加入1.0 mL酒石酸钾钠溶液,摇匀再加入纳氏试剂2.0 mL,摇匀。放置10 min后,在波长420 nm下,用10 mm比色皿,以水做参比,测量吸光度[5]。
称约5 g试样(精确到0.000 1 g),用水溶解,定容到250 mL。吸取2 mL样品溶液于50 mL比色管中,其余步骤与1.2.2加水至标线开始,同上。
2 结果分析
2.1 方法检出限
依据标准HJ 168-2020中规定,滴定法检出限一般根据所用的滴定管产生的最小液滴的体积来计算[8],计算公式为:
(1)
式中:MDL——方法检出限;λ——被测组分与滴定液的摩尔比;c——盐酸标准滴定溶液的浓度,mol/L;V——滴定管所产生的最小液滴体积,mL;M——铵的摩尔质量,g/mol;m——称取样品的质量,g;k——当为一次滴定时,k=1;当为返滴定或间接滴定时,k=2。
该试验为酸碱中和滴定,因此,λ=1,k=1,c=0.05 mol/L,V=0.05 mL,M=18.038 5 g/mol,m=10 g,计算出方法检出限MDL=0.000 45%。
2.2 准确度
选取3个含有不同铵含量的氯化钾样品(试样1、试样2、试样3),按照样品中铵含量的高低加入标准物质,试样1和试样2加质量浓度为1 000 μg/mL的铵标准溶液,试样3加质量浓度为100 μg/mL的铵标准溶液,每个样品加3个不同含量的标准物质,分别加入:试样1,5 mL、10 mL和15 mL;试样2,2.5 mL、5 mL、10 mL;试样3,1 mL、2 mL、3 mL,对应的含量见表1。样品实际测定值为X0,加入标准物质后试样中铵含量的测定值为X1,回收率P按式(2)计算:
(2)
式中:P——回收率,%;m——试样的质量,g;X0——加标前样品中铵的含量,mg;X1——加入标准物质后试样中铵的含量,mg;X2——加入铵标准物质含量,mg。
由表1可知,试样1加标回收率分别为103.74%、104.96%和104.88%,试样2加标回收率分别为93.50%、102.81%和100.40%,试样3加标回收率分别为97.46%、93.70%和92.39%,不同含量样品,依次增加加入标准物质的含量,铵的回收率在90%~105%之间,有较高的准确度。
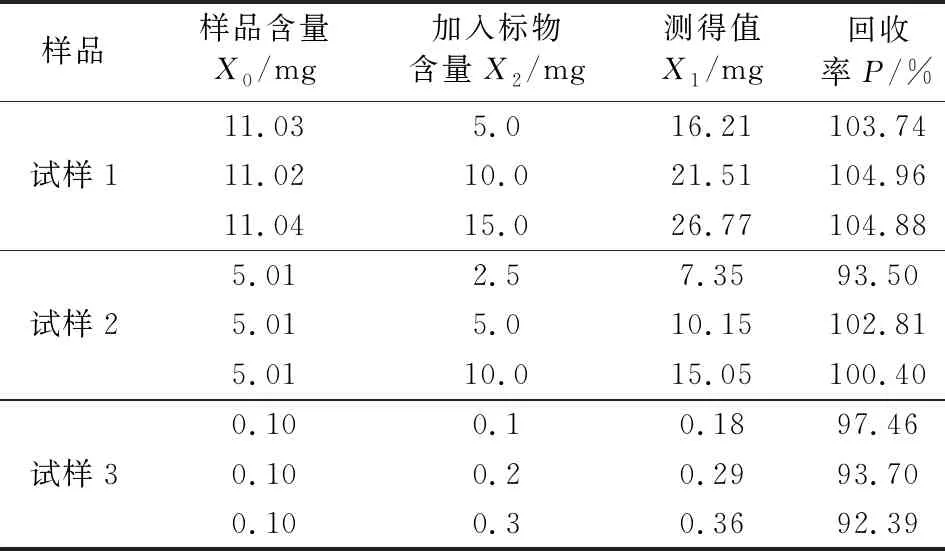
表1 氯化钾中铵含量的回收率试验结果Tab.1 Test results of recovery rate of ammonium content in KCl
2.3 精密度
选用5个氯化钾样品,粉碎后过0.5 mm试验孔径筛,按照1.2.1方法对每个样品进行6次平行测定,试验结果结果见表2。从表2可知,5个样品的相对标准偏差(RSD)分别为0.59%、1.80%、0.86%、1.77%和9.10%,最大极差分别为0.002%、0.002%、0.001%、0.002%、0.000 2%。实验结果表明,蒸馏后滴定法测定氯化钾中铵含量离散程度小,精密度高。

表2 氯化钾中铵含量的重复性Tab.2 Repeatability of ammonium content in KCl %
2.4 方法比对
取同一氯化钾样品,利用蒸馏后滴定法和纳氏试剂分光光度法分别进行6次平行测定。对6次检验结果进行F检验[9]和t检验[10]。
利用两种方法对同一待测样品中的铵含量进行F检验,结果如表3所示。可以看出,P(sig)值=0.765>0.05,故选择看“假设方差相等”一行结果。t=1.754,df=10,P(sig)值=0.110>0.05,说明两组试验结果数据差异不显著,即蒸馏后滴定法和纳氏试剂分光光度法对铵的检测差异不明显。因此,蒸馏后滴定法和纳氏试剂分光光度法检测铵含量具有较好的一致性,可以用蒸馏后滴定法测定氯化钾中铵含量。纳氏试剂分光光度法是将氯化钾溶解后蒸馏,然后再使用纳氏试剂显色,分光光度计测定[9],此方法步骤较繁琐、耗时长、且使用二氯化汞和碘化汞剧毒物质,对人体伤害性极大。而此方法步骤简便、不使用剧毒物质,安全性高,方法精密度和准确度高。因此,在实际检测中蒸馏后滴定法较实用、高效。

表3 独立样本检验Tab.3 Independent sample test
3 结论
文章采用蒸馏后滴定法测定氯化钾中的铵含量,并且与纳氏试剂分光光度法进行比较,通过测定同一样品对结果数据进行F检验和t检验,表明两种方法差异不明显。蒸馏后滴定法进行了方法检出限、准确度和精密度试验,结果表明,该方法检出限为0.000 45%,加标回收率在90%~105%之间,最大相对标准偏差<10%,最大极差≤0.002%,该方法具有较高的准确度与精密度。相比较于纳氏试剂分光光度法,具有操作简便、耗时短、安全性高等特点,因此,在化工行业以及以氯化钾为原料生产其他产品的行业中具有较大的应用潜力。
