Nitroxy-and azidomethyl azofurazans as advanced energetic materials
2022-08-30AlekseiSheremetevSvetlnMelnikovElizvetKokrevRuslnNekrutenkoKirillStrizhenkoKyrillYuSuponitskyThnhDtPhmAllPivkinVlerySinitskii
Aleksei B.Sheremetev ,Svetln F.Mel'nikov ,Elizvet S.Kokrev ,Rusln E.Nekrutenko , ,Kirill V.Strizhenko ,Kyrill Yu Suponitsky ,Thnh Dt Phm ,All N.Pivkin ,Vlery P.Sinitskii
a Zelinsky Institute of Organic Chemistry,Russian Academy of Sciences,47 Leninsky Prosp,Moscow,119991,Russia
b Saint Petersburg State Institute of Technology,26 Moskovskii Pr,St Petersburg,190013,Russia
c Mendeleev University of Chemical Technology,Moscow,125047,Russia
d N.N.Semenov Federal Research Center for Chemical Physics,Russian Academy of Sciences,4 Kosygina Str,119991,Moscow,Russia
Keywords:Azofurazan Azide Nitrate ester X-ray Combustion
ABSTRACT Progress in the rocket industry is only possible on the basis of new,higher performance and more environmentally friendly materials compared to up-to-date propellant ingredients for liquid,solid,gelled and hybrid propellant systems.In this work,synthetic methods have been developed for the preparation of new energetic azofurazans bearing nitroxymethyl or azidomethyl groups.All prepared compounds were fully characterized by multinuclear NMR and IR spectroscopies,as well as elemental analyses.An analysis of the structural features based on the X-ray single-crystal diffraction made it possible to discuss their influence on the densities of the azofurazans of this study.Thermal decomposition and combustion of nitroxymethyl and azidomethyl azofurazans were studied using a number of complementary experimental techniques,namely thermogravimetry,differential scanning calorimetry,manometry,microthermocouple measurements in the combustion wave.The structural and physical characteristics of these new energetic analogues illustrate the extent to which the nature of the explosophoric groups can be used to tune the performace of the azofurazan framework.These azofurazans possess positive calculated enthalpy of formation and are promising candidates for new environmentally friendly energetic materials.
The traditional strategy for newly developed explosives and propellant ingredients is to combine various explosophoric groups(-NO,-NNO,-ONO,-N,-ClO,etc.)with diverse frameworks in a target molecule [1a-d].The current trend is to use nitrogen heterocycles as frameworks [2a-i].
Nitrate esters are a famous class of available compounds that are known for their widespread use as components of binders for energetic materials for various purposes[3].Of these,cellulose nitrate and glycerol trinitrate are well-known nitrate esters that were first produced more than a century and a half ago and are widely used to this day.A wide variety of new compounds bearing a nitroxyalkyl group have been synthesized and some of them have found military use [3],or have demonstrated therapeutic potential as drugs [4a,b].Despite a flurry of synthetic activity in area of alkyl nitrates,there have been relatively few reports of energetic nitroxymethylazoles.Indeed,only rare furazans [5a-c],isoxazoles[6a,b],1,2,4-oxadiazoles [7],1,2,3-[8a,b] and 1,2,4-triazoles [9]bearing a C-attached nitroxymethyl group are described in the literature as ingredients of energetic materials (Fig.1).Several(nitroxymethyl)furoxans have also been synthesized,but studied only as vasodilators [10a,b].
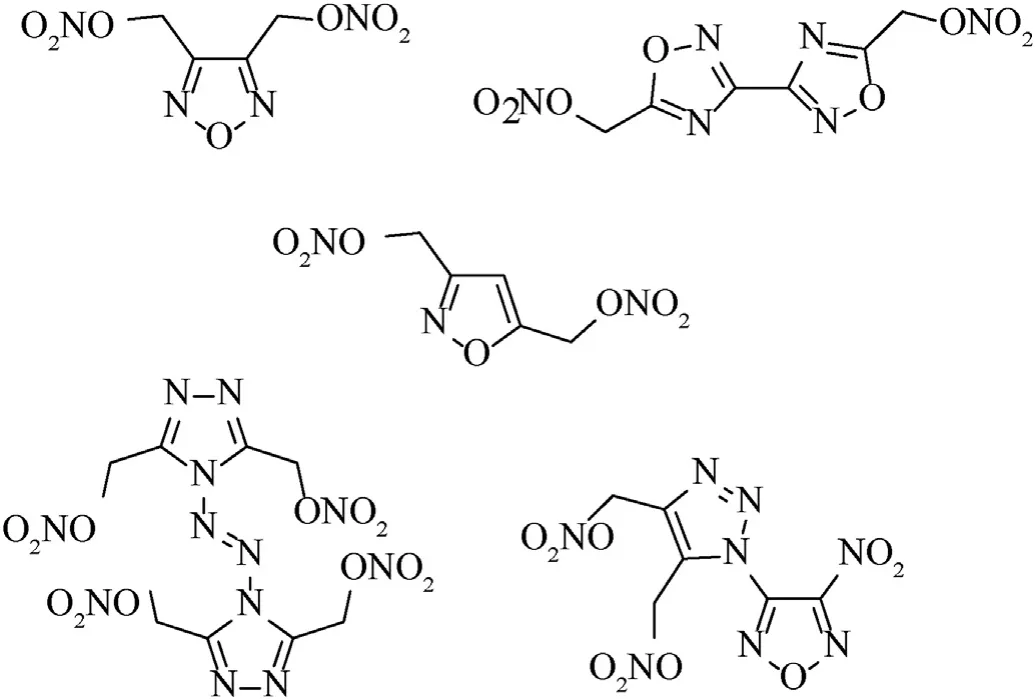
Fig.1.(Nitroxymethyl)azoles -energetic plasticizing ingredients of propellant formulations or melt-castable eutectic ingredients of energetic materials.
Over the past five decades the chemistry of furazans (1,2,5-oxadiazoles) has been investigated intensively [11a-d] and,among other things,has led to the creation of many highperformance energetic furazan-based compounds [12a-e].On the other hand,a large number of promising energetic compounds have been developed based on the azoazole frameworks [13].Among them,azofurazans have long been known as attractive ingredients with highest enthalpies of formation for use in explosives and propellants [14].In our quest to develop new synthetic routes to novel energetic azofurazan combinations,we synthesized 3-amino-4-hydroxymethylfurazan (1) as a key building block for the creation hitherto unknown nitroxymethyl and azidomethyl azofurazans and evaluated their physical,structural,thermochemical,and energetic properties.Herein,the results of our research are presented.
1.Results and discussion
The alcohol 1 has previously been prepared by the reduction of propyl ester 3-aminofurazancarbonic acid with LiAlHin THF[15].Taking into account the inconvenience of using LiAlH,sodium borohydride was used as an undemanding reducing reagent to transform 3-aminofurazancarbonic acid methyl ester (2)[16] into 3-amino-4-hydroxymethylfurazan (1).As shown in Scheme 1,treatment of readily available ester 2 with sodium borohydride in MeOH afforded alcohol 1 in~80% yield.This reaction is easily scalable,and using the pathway in Scheme 1,we were able to produce alcohol 1 on a kilogram scale.The furazan 1 is a bifunctional heterocyclic building block as it has two reactive centers,amino and hydroxymethyl groups,and the chemo-and regioselectivity of their reactions under various conditions is,therefore,of undoubted interest.
The oxidative dimerization of(het)aromatic amines to yield azo compounds is a time-tested synthetic tool of enormous importance.In particular,azofurazans are synthesized from the corresponding aminofurazans.There are a number of protocols available for this transformation involving the use of various oxidants such as KMnO/H[17a-h],CrO/AcOH[18],(NH)SO[19],NaOCl or NaOBr[20a-c],nitronium tetrafluoroborate[21],Br/HO[22],a variety of organic reagents(dibromoisocyanuric acid[23a,b]and trichloroisocyanuric acid [24a,b]).The electrochemical oxidation of aminofurazans on nickel oxyhydroxide anode is also possible [25].However,both amino and hydroxymethyl groups of compound 1 can participate in the oxidation reaction.
Really,screening of various conditions for oxidation 1 showed that almost each of the above oxidants provides the formation of an azo compound,which is usually accompanied by several byproducts,demonstrating the participation of the hydroxymethyl group in the oxidation.However,we found that available and inexpensive NaOCl,as an aqueous solution,was the most effective and selective for converting amine 1 to the desired azo diol 3 in~85% yield,with this reaction also being readily amenable to scaleup.It was found that the nitration of diol 3 proceeds smoothly both in 100% HNO/AcO or 100% HNO/98% HSOand in pure 100% HNOto provide dinitrate ester 4 in high yields (Scheme 1).The scalability of the foregoing nitration protocols was then examined.It is noteworthy that on a large scale(0.1 mol),only pure nitric acid showed good batch-to-batch reproducibility,consistent yield and product purity.Moreover,in this case,the regeneration of the spent acid is easier than when using nitrating mixtures.
For comparison,azofurazan bearing azidomethyl groups,diazide 8,was also prepared.Three approaches were used to synthesize 8 as outlined in Scheme 2.In an initial approach,alcohol 1 was converted to the corresponding chloromethyl compound 5,which was subjected to azidation with NaNin the presence of KI to obtain 3-amino-azidomethylfurazan (6).Oxidation of the amino group of 6 with KMnOin hydrochloric acid produced target diazide 8 in good total yield.
In another approach,the azidation and oxidation steps were performed in reverse order,first obtaining the azo compound 7 and then replacing the chlorine atoms in it with azido groups.The isolated yield of pure product 8,calculated on the basis of starting 1,was generally in the 60-70% range,that is,similar to the first approach.
An alternative approach was to convert dinitrate 4 to diazide 8.As a number of groups had reported the nucleophilic displacement of nitroxy group with azide ion to generate the corresponding organic azides[26a-f],these conditions were applied to dinitrate 4 in an attempt to produce the diazide 8.Indeed,diazide 8 was obtained by the azidation reaction of dinitrate 4 with sodium azide in the presence of urea in a mixture of DMF and water (5:1) in good yield (Scheme 2).
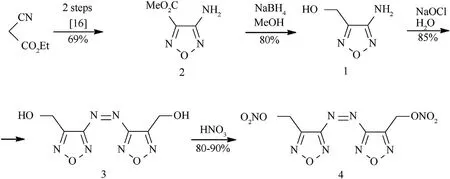
Scheme 1.Synthesis of dinitrate ester 4.
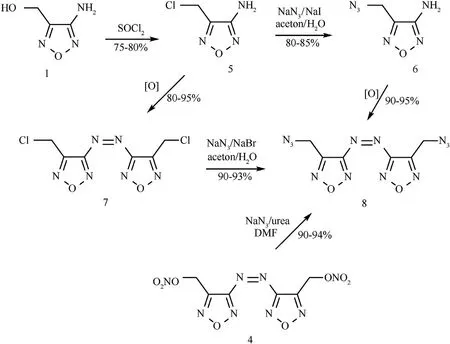
Scheme 2.Synthesis of diazide 8.
We believe that all three approaches are valuable,depending on the availability of the required reagents,providing similar yields based on the starting alcohol 1.
The structures of the all compounds were confirmed by NMR,and IR spectroscopy,and elemental analysis,and for azofurazans 4,7,and 8 also corroborated by X-ray crystallography.
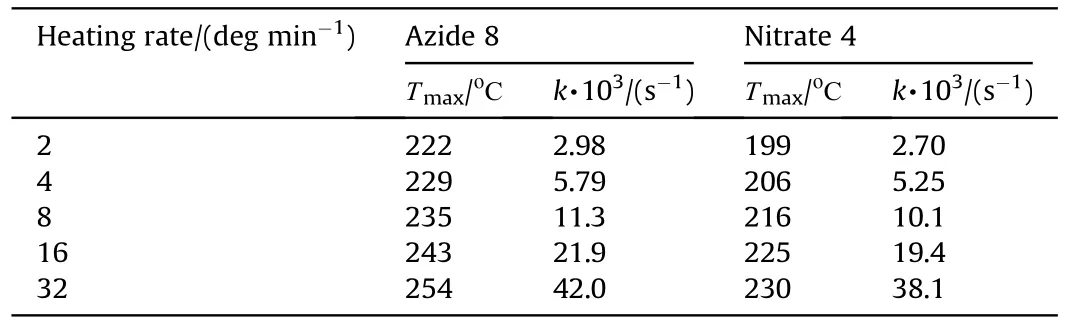
Yellow,monoclinic crystals of nitare 4,chloride 7 and azide 8 were obtained by slow evaporation of CCl,CHCland CHClsolutions at ambient temperature,and their solid-state structures were determined by single crystal X-ray diffraction(for details,see ESI).
All compounds crystallize in P2/c space group,and their molecules are centrosymmetrical and occupy a special position in the unit cell.An asymmetric unit cell contains half of molecule for 7 and 8 and two halves for 4.As can be seen in Fig.2,the molecular backbone of all studied compounds is nearly planar and characterized by the usual ap-ap-ap configuration [27,28].However,the orientations of substituents CHX(X=Cl,N,ONO)with respect to rotation about the C2-C3 bond are distinctly different.In both 8 and 4,the substituents are directed away from each other,and the N4 and O2 atoms are located nearly in the molecular plane.In compound 7,the chlorine atoms are significantly outside the molecular plane.The torsion angles that determine the molecular conformation are given in Table 1.To find an explanation for this,we performed a quantum chemical calculation using the M052X functional with a triple-zeta basis,which previously proved to be reliable for assessing the structural and energy properties of heterocyclic compounds [29-32].
For all compounds,two minima were observed,namely,global and local.In all cases,one minimum corresponds to a nearly planar orientation of the substituents;for 7 and 8,it is local,and for 4 it is global (Table 1).Therefore experimental conformation of 7 corresponds to a global minimum,conformation of 8 corresponds to a local minimum,and experimental structure of 4 is between two minima.As we will see below,all these findings are closely related to crystal packing effects.
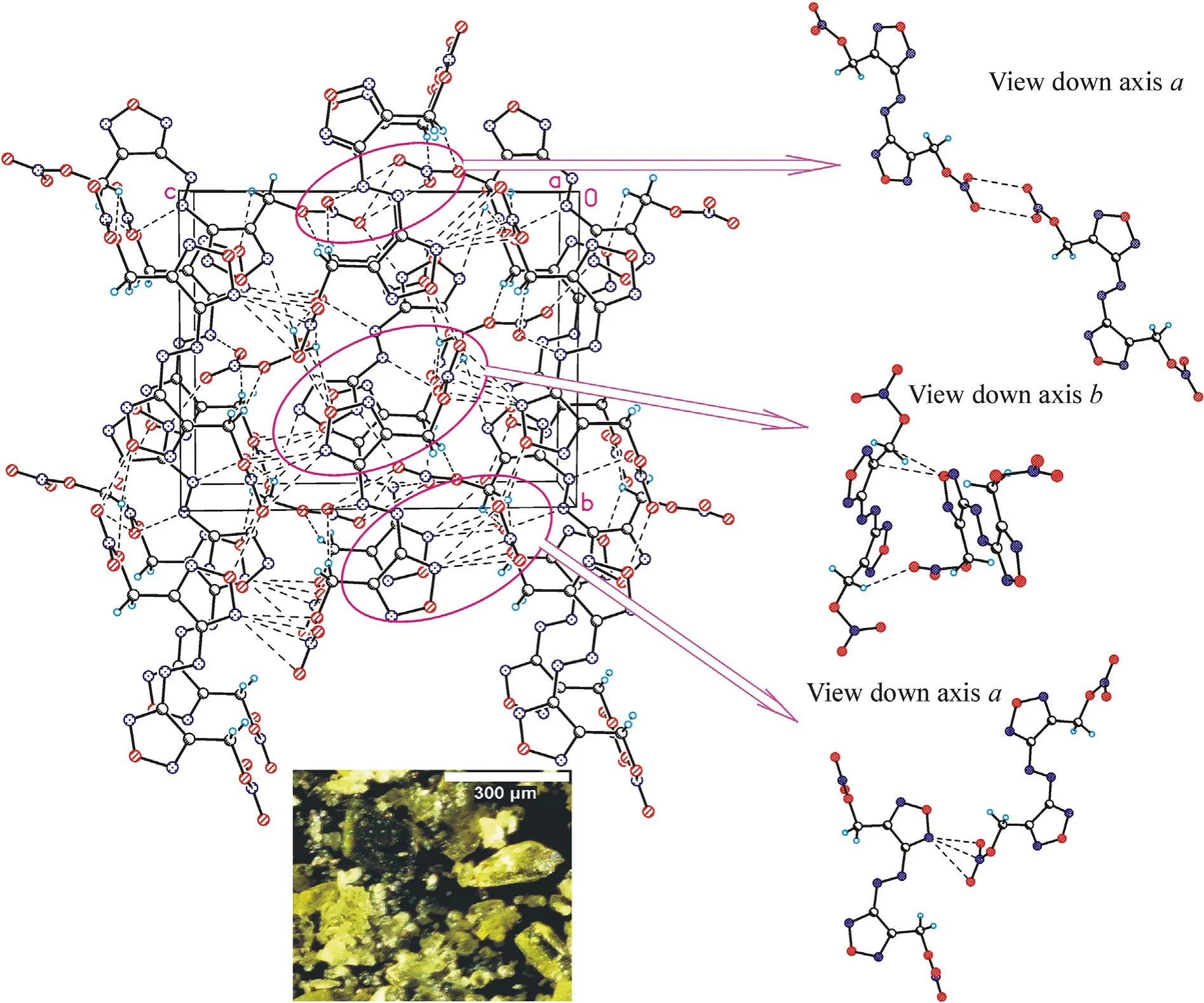
Crystal structures for the three azofurazans show that specific intermolecular contacts formed by N and O atoms of the azido-and nitroxy groups as well as furazan rings are powerful tools for dense crystal engineering.The densities of the crystal structures of chloride 7 and azide 8 are approximately the same (1.742 and 1.739 g/cmat 100 K),while the density of nitrate 4 is much higher(1.816 g/cm).This can be explained by the degree of participation of the Xgroup in intermolecular bonding.As clearly shown in Fig.3 (also see Table 1S in ESI),the chlorine atom is not involved into the system of intermolecular close contacts.However,chlorine is a heavy atom compared to other second-row atoms,and the observed density of compound 7 is merely due to its presence in the molecule.As can be seen in Fig.4 (also see Table 2S in ESI),azido groups of 8 participate in four O … π interactions between the terminal nitrogen atom of the azido group and oxygens from the furazan ring,as well as in four pairwise bonded stacking interactions,two of which are the strongest in the crystal 8 and are probably responsible for the planar molecular conformation.In general,the intermolecular bonding in azide 8 is much more pronounced than in chloride 7,mainly due to the activity of the Ngroup(for comparison,see ESI,Tables 1S and 2S).This leads to the fact that the density of 8 is almost the same as that of chloride 7.
In nitrate 4,the ONOgroups are involved in all pair interactions as shown in Fig.5(see ESI,Tables 3S and 4S).The O3 and O4 atoms are out of the plane of the framework (Fig.1),and instead of stacking interactions observed in compounds 7 and 8,crystal structure of 4 is stabilized by low-directional O(N) … π and C-H…O(N) contacts.Moreover,all molecular pairs in 4 are linked via specific intermolecular interactions in addition to ordinary vander-Waals contacts.As a result,crystal packing of nitrate 4 is the densest among the compounds in this study.
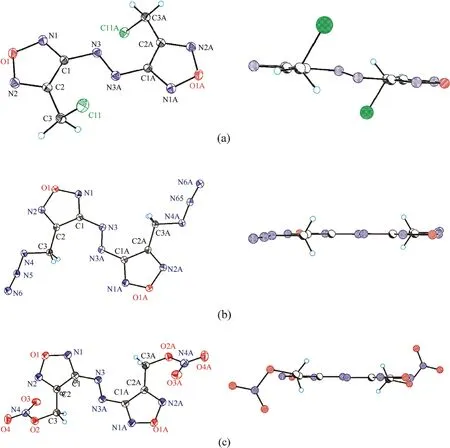
Fig.2.(Left) Thermal ellipsoid plot (50%) and labeling scheme for compounds 7 (a),8 (b) and 4 (c).(Right) Lateral view of 7,8 and 4.
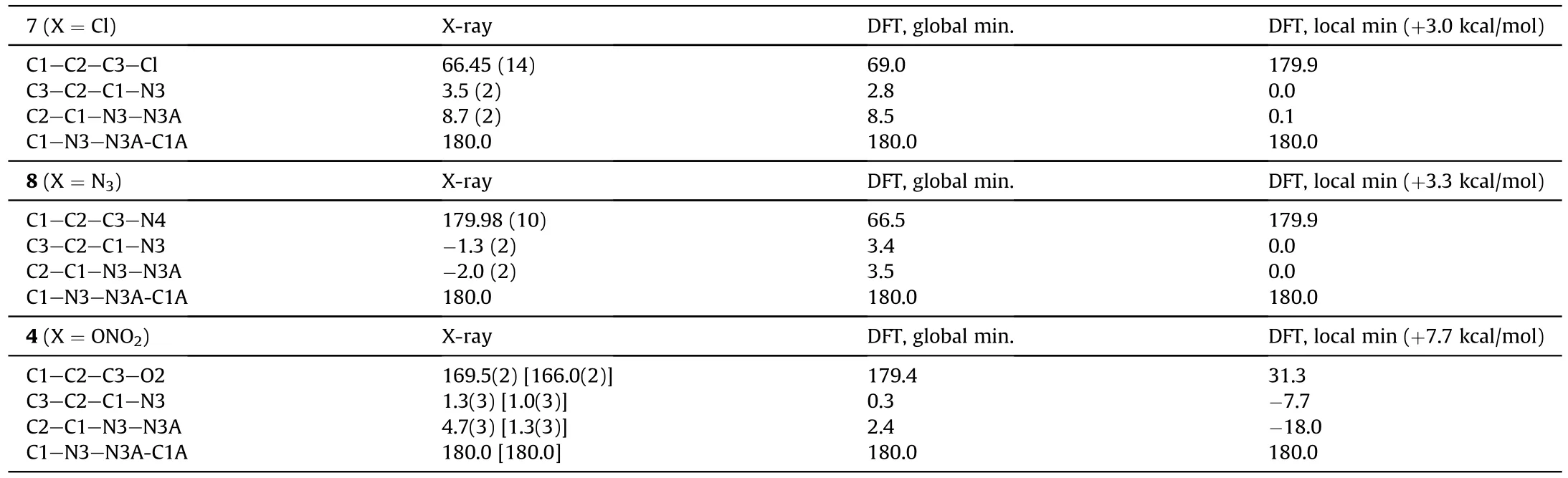
Table 1.Selected torsion angles defining molecular conformation of compounds 7,8 and 4.Theory vs.X-ray experiment.
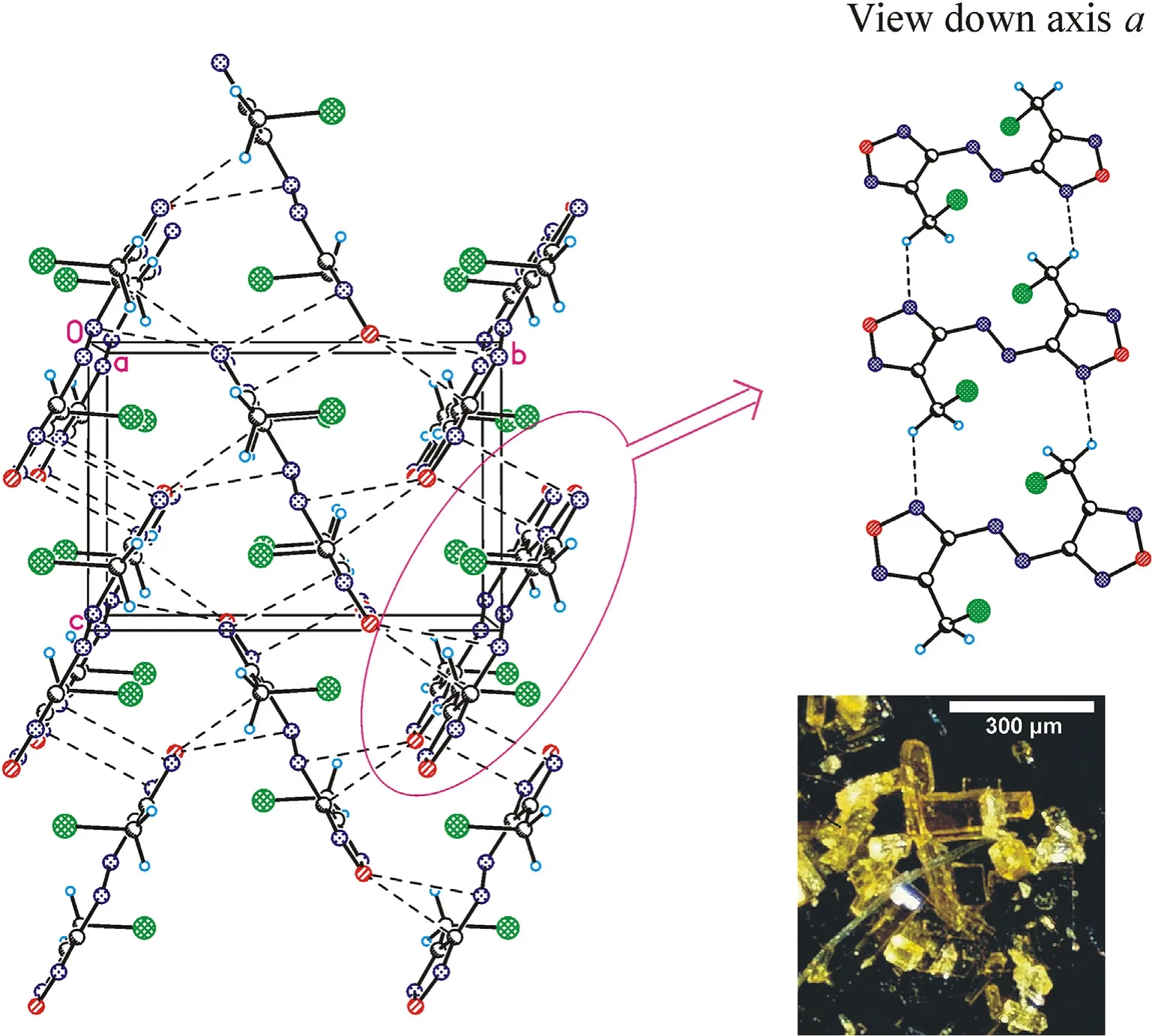
Fig.3.Crystal packing fragment of chloride 7: projection onto the bc plane.(Left) Stacking interactions linked molecules into chains along the c axis;O … π interactions linked chains into layers parallel to the bc plane.(Right) C-H…N contacts connect the layers in a 3-D structure (see ESI,Table 1S).Photo: crystal appearance.
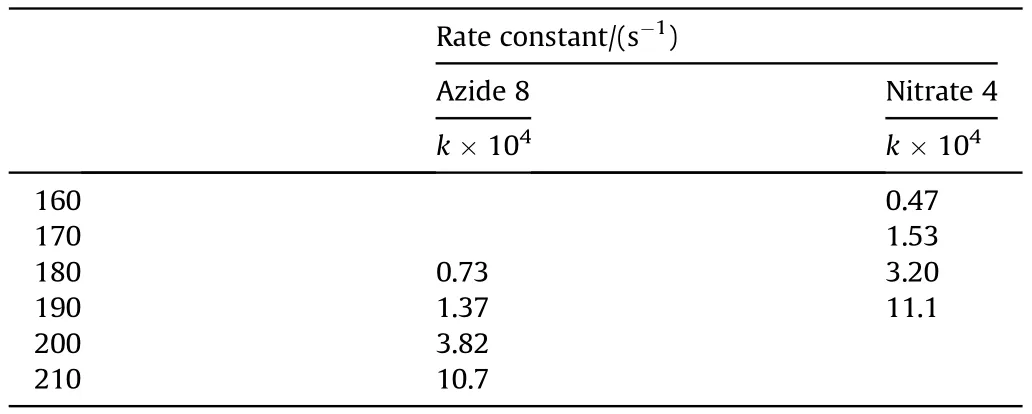
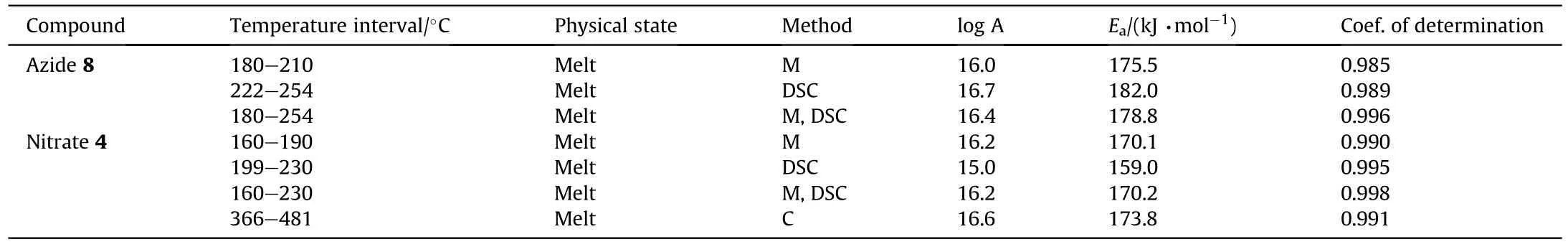
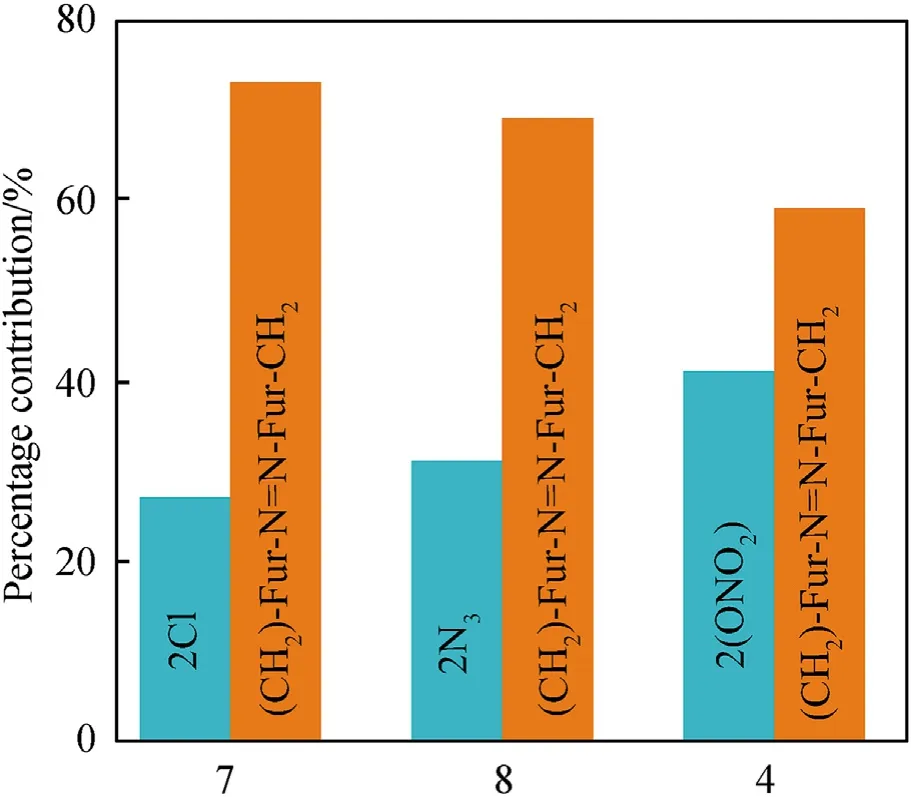
A direct comparison of the contributions of the Cl,Nand ONOgroups into the molecular and crystal density can be quantified in terms of the recently proposed Δ-based densification approach[33-36].According to this method (details of which are given in ESI),an isolated molecule placed in a crystal becomes denser due to intermolecular interactions.The difference between the molecular densities in the crystal and in the isolated state (Δcriterion)Δ=d-dwould define tightness of the crystal packing.The Δcriterion can be calculated both for the whole molecule and for any molecular subunit.Molecular and crystal densities of compounds 7,8 and 4 and their subunits are summarized in Table 2.
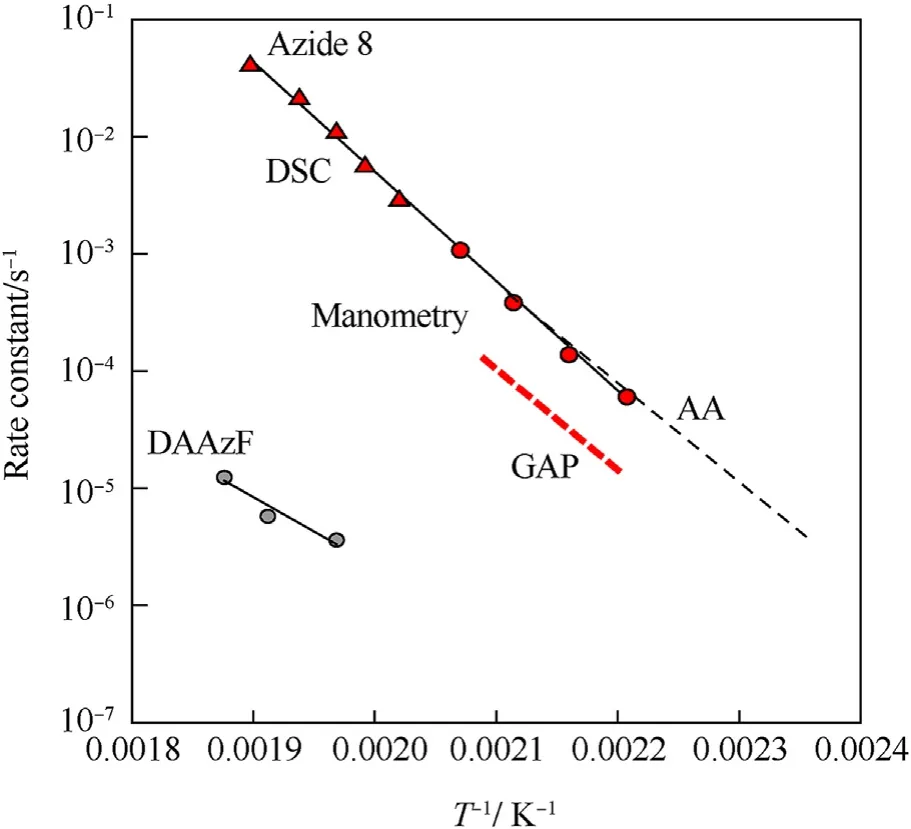
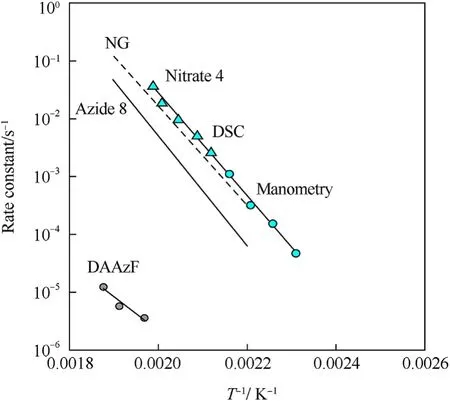
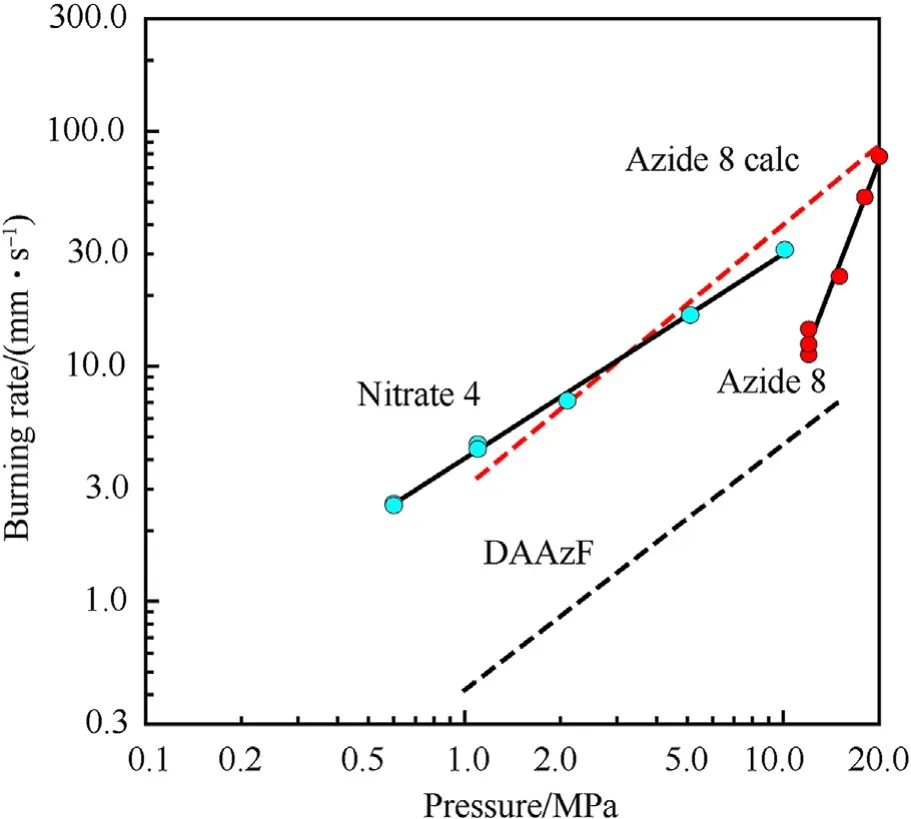
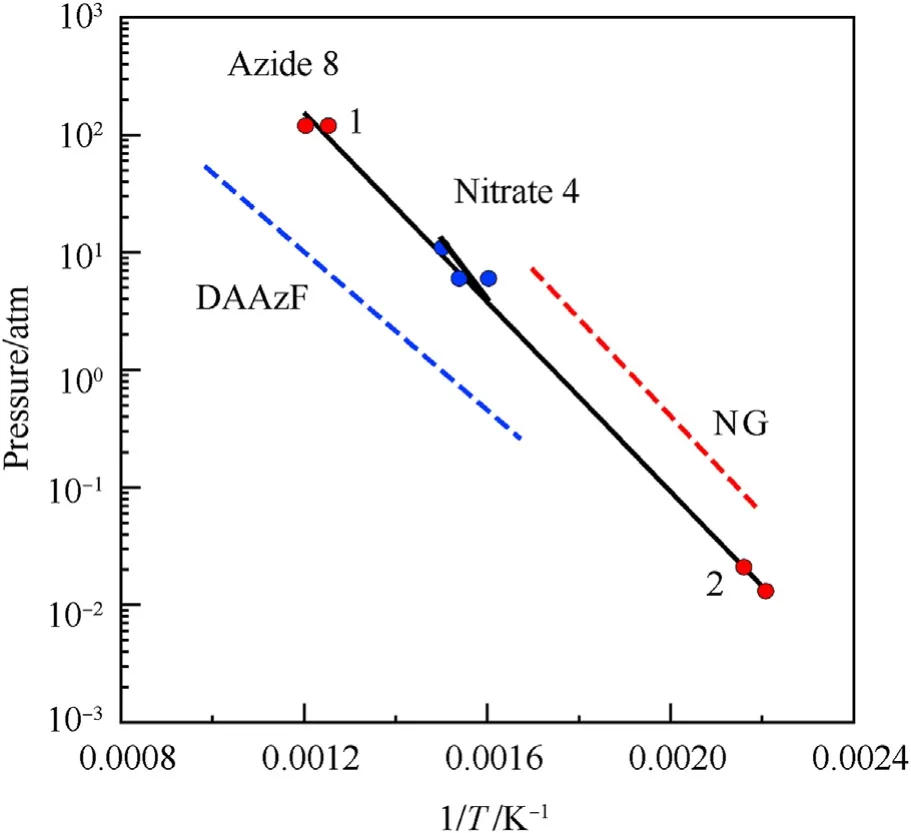
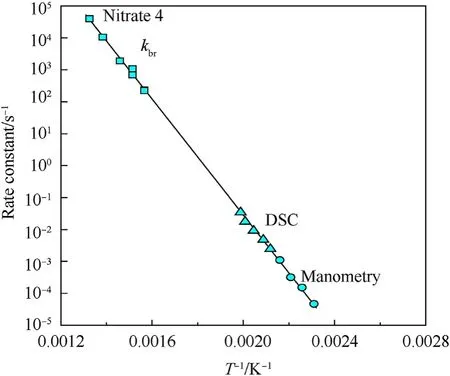
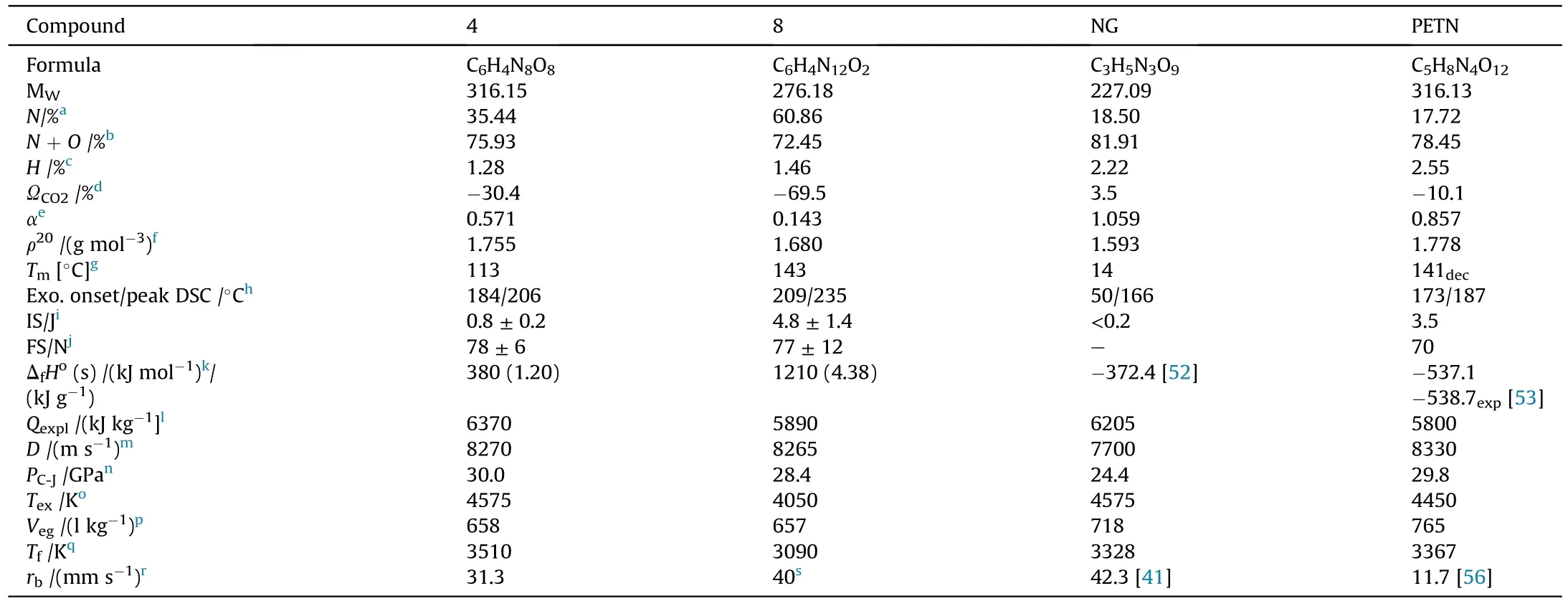
It can be seen that the results of Table 2 are in good agreement with the above conclusions,and also provide some additional information.The chlorine atom contributes to the density mainly due to its mass,and much weaker due to its densification(Δ=0.389 g/cm),which is the smallest among the compounds in this study.The azido group densifies much more tightly(Δ=0.449 g/cm)compared to chlorine,that is to some extent compensation for its low molecular density(d=1.233 g/cm).It is interesting to note that the ONOgroup,built from atoms of the second row,turns out to be a denser unit (d=1.555 g/cm)compared to chlorine (d=1.408 g/cm) due to its compact structure.It is also seen that the ONOgroup extensively participates in intermolecular bonding and has the greatest value Δ(0.457 g/cm).Based on the densification analysis,we can estimate the percentage contribution of the X substituent and the rest of the molecule to the crystal density (see ESI for details of the calculation).Fig.6 clearly demonstrates the contribution of the X substituent to the crystal density,which grows in the following order:Cl The thermal properties of nitrate 4 and azide 8 were investigated with differential scanning calorimetry (DSC,scanning at 5C/min,see the Supporting Information).Nitrate 4 has a melting point of 113C with an enthalpy of melting of 31.6 kJ/mol(110 kJ/kg),while azide 8 melts at 143C with an enthalpy of melting of 30.4 kJ/mol (100 kJ/kg).Compound 4 begins to decompose at 184C with a heat effect equivalent to~393 kJ/mol(1423 kJ/kg),and compound 8 at 209C with a heat effect of~452 kJ/mol(1430 kJ/kg).That is,the thermal safety interval(Δ=T-T)for compounds 4 and 8 is 71 and 66С,respectively.It is important to note that both compounds are thermally more stable than a benchmark compound such as pentaerythrityl tetranitrate (PETN,Tis 156C). Under further heating,for 8 another broad peak of heat release is recorded with a maximum at 296C,and the total thermal effect is 723 kJ/mol (2622 kJ/kg).For 4,the second peak is observed at 262C,but it is very weak. Fig.4.(Left)Crystal packing fragment of azide 8:projection onto the ac plane.Hydrogen atoms are not involved into the close contacts system,so they have been omitted for clarity.(Right) O(N) … π and stacking interactions (also see ESI,Table 2S).Photo: crystal appearance. As a result of decomposition at different heating rates,the rate constants of thermal decomposition of both compounds were determined by the Kissinger method [37],assuming a first-order reaction,and are given in Table 3. The experiments on the decomposition of compounds 4 and 8 under isothermal conditions were carried out in thin-walled glass manometers of the compensation type(the Bourdon glass gauge). Gas evolution during decomposition of compound 8 (m/V~ 10g/cm) was investigated in the temperature range of 180-210C (see ESI,Figure S3).It has been shown that as the temperature rises from 190 to 210C,gas evolution increases from 200 to 270 cm/g (or from 2.5 to 3.3 mol of gases per mole of the starting 8).This indicates the presence of a compound with high volatility in the decomposition products,and the increase in gas evolution is due to its vapor pressure.This assumption is confirmed by a large amount of condensable products(15% or~0.6 mol/mol)upon cooling the gases to room temperature.After the decomposition of 8,a solid dark brown residue remained in the Bourdon flask.This residue is practically insoluble in common solvents.The IR spectrum of the residue showed the indicative loss of the azide bands at ca.2100 cm.In this IR spectrum,a characteristic broad band at ca.3420 cmcan be attributed to a NH-group,while absorption band for the C═N bond was observed at ca.1620 cm. Unlike azide 8,for nitrate 4 the maximum gas evolution during decomposition does not increase with increasing temperature and is 420-430 cm/g or 5.9-6.1 mol of gases per mole of the starting 4(see ESI,Figure S4).This indicates a deeper decomposition of the initial molecule at this stage.The amount of condensed products when gases are cooled to room temperature is 35% or~2 mol/mol.The residue after decomposition is a yellow solid,in the IR spectrum of which there are broad absorption bands in the region of OH group(3200-3600 cm) and C═N bonds (~1610 cm). The rate of gas evolution during the decomposition of azide 4 can be described by the first order to the depth of decomposition of 40-50%.The obtained rate constants (Table 4) are in good agreement with the constants from non-isothermal conditions(Fig.7).A similar trend is observed for nitrate 4(Fig.8)Decomposition kinetic parameters determined by various methods for compounds 4 and 8 are given in Table 4 (see Table 5). Table 2.Densities (g/cm3) and ΔOED (g/cm3) values of molecular subunits of compounds 7,8 and 4.a Table 3.DSC analysis data for compounds of this study. Table 4.The isothermal decomposition results of compound 4 and 8. Table 5.The Arrhenius parameters of thermal decomposition of compounds 4 and 8 obtained from DSC technique,isothermal manometric kinetics (denoted as M),and combustion measurements(C label). As can be seen from Fig.7,the decomposition kinetics of azide 8 and azidoacetone(AA)are nearly the same.In contrast,compound 8 degrades~3 orders of magnitude faster than DAAzF,which begins to decompose with the loss of the Nmolecule from the azo group.This is due to the fact that at the first stage only the azido group decomposes,while the azo group and furazan rings remain intact.This is supported by the IR spectrum of the residue formed during decomposition,where the absorption of the azide group disappears and the absorption in the region of the C=N bond is enhanced,as well as the release of 2 mol of non-condensable gases (N) (upon decomposition under isothermal conditions).Calculation of the heat effect of this reaction gives 1485 kJ/kg,which is in good agreement with the heat release under DSC conditions(1430 kJ/kg).The azo group and the furazan ring decompose at higher temperatures (~300C) in the following stages. Fig.5.(Left)Crystal packing fragment of nitrate 4:projection onto the ac plane(Right)O(N)…π,C-H…O and O…O interactions(also see ESI,Table 2S).Photo:crystal appearance. Fig.8 demonstrates that the kinetics of decomposition of nitrate 4 and nitroglycerin are practically the same,but 3 orders of magnitude faster than DAAzF.Obviously,decomposition of the nitro ester group is responsible for this process.The cleavage of the NOgroup from the nitroxymethyl group produces an intermediate alkoxyl radical,which further decomposes with the release of formaldehyde and the formation of a polymer from the remaining azofurazan backbone.Next,redox recombination of formaldehyde with nitrogen oxides leads to the formation of 6 mol of gases,of which 2 mol are condensing (HO): Azide 8 in the form of charges with a density of 1.55 g/cm(92% TMD) pressed into acrylic tubes with an inner diameter of 4 mm does not burn up to a pressure of 12 MPa.At higher pressures,the azide burns with a glowing flame.The burning rate in the range of 12-20 MPa is described by a law with an unusually high pressure index r(mm/s)=0.002 P(Fig.9). Its analog,nitrate 4,in charges with a density of 1.58 g/cm(90% TMD) begins to burn from 0.6 MPa.The combustion rate in the range of 0.6-10 MPa is described by the quite usual law r(mm/s)=4.05 P(Fig.9).The combustion rate of nitrate 4 is higher than that of DAAzF[39]and slightly lower than that of nitroglycerin(NG)[41]. The absence of combustion in the range of 0.1-12 MPa and a large index in the combustion law of azide 8 indicates the presence of some kind of combustion instability.This is unusual,since,according to DSC,azide 8 decomposes as a typical organic azide,and the heat effect of azide groups destruction is quite sufficient to ensure the combustion process.This behavior could be explained by thermocouple studies of the combustion wave. Fig.6.Comparison of the percentage contributions of substituents Cl,N3,ONO2 and the framework,CH2-Fur-N═N-Fur-CH2,into the crystal density. The temperature distribution in the combustion wave of azide 8 was obtained using fine tungsten-rhenium thermocouples only at a pressure of 12 MPa (see ESI,Figure S5).At higher pressures,the burning rate is very high,so the thermocouple technique is not applicable.The temperature profiles show a one-stage flame structure and can be used to establish the surface temperature.The maximum measured flame temperature is 2015 ± 25 K,which is significantly lower than the calculated adiabatic temperature(3090 K).Earlier it was shown [39] that in the absence of an oxidizer,the furazan ring does not release all the energy stored in it due to the formation of endothermic products bearing nitrile group.A similar trend is observed during the combustion of azides under oxygen deficiency [42].If,when calculating the adiabatic temperature of compound 8,4 mol of HCN are fixed in the combustion products (2 mol from azide groups and 2 mol from furazan rings),then this temperature will drop to a value of 2510 K.The overestimation of the calculated temperature over the experimental one can be explained by the losses of the thermocouple to radiation,which is usually at temperatures above 2000 K [43]. Fig.7.Comparison of the decomposition kinetics of azide 8 and glycidyl azide polymer(GAP),azidoacetone (AA) [38] and diaminoazofurazan (DAAzF) [39]. Above data at one pressure does not allow determining the dependence of surface temperature on pressure.However,if thermocouple data on surface temperatures at 12 MPa are combined with the initial vapor pressure in the Bourdon manometer (in the first 2-4 min)(Fig.10),then such the dependence in a wide range of pressures can be described by eq.(1): According to Eq.(1),the heat of vaporization (L) of azide 8 is 77 kJ/mol (280 J/g). The temperature gradient over the combustion surface at 12 MPa is φ=1.74 × 10K/cm.Using the thermal conductivity coefficient λ=8.8×10J/cm×K calculated by the Real Code[44],the heat flux,q,from the gas phase to the surface is 155 J/cms,and taking into account the burning rate,the amount of heat coming from the gas phase is Q=q/m=84 J/g.This heat is not enough even for the evaporation of azide 8 (280 J/g),let alone for heating this compound to the surface temperature. On the other hand,the heat released in the condensed phase is sufficient for heating the compound to the surface temperature and melting.Indeed,according to DSC (see above),1430 J/g is released during decomposition of azide 8,and 100 kJ/kg is spent on melting,the rest of the heat could be used for heating.At the surface temperature for compound 8,the heat capacity coefficient is c=1.66 J/g K(calculated using the Real Code[44]).Hence it follows that this heat is sufficient for heating up to a temperature of 1074 K(1430-100)/1.66+273),while even at a pressure of 15 MPa,the recorded surface temperature,T,is not more than 815 K. Fig.8.Comparison of the decomposition kinetics of nitrate 4 (triangles -DSC,dots -manometry) and nitroglycerin (NG) [40] and DAAzF. Fig.9.Comparison of the combustion rates of nitrate 4,azide 8,DAAzF and NG.The calculated combustion rate of azide 8 is also given. What is the reason for the absence of combustion in the range of 0.1-11 MPa? Knowing the dependence of the surface temperature on pressure,and having the experimental heat of decomposition(DSC)and the calculated thermophysical parameters,it is possible to estimate the combustion rate in a wide pressure range by the combustion model with a leading reaction in the condensed phase [45] using Eq.(2): Fig.10.Comparison of the vapor pressure of azide 8(1-surface temperature,2-initial pressure in the manometer),nitrate 4,DAAzF and NG. where c,ρ,χ are the heat capacity,density and thermal diffusivity of the condensed phase,T,Q and Lare the surface temperature,the heat effect and the heat of fusion,E and A are the activation energy and the preexpotential factor of the leading reaction in the condensed phase. Comparison of the experimental and calculated dependences is shown in Fig.9.As it is seen,according to the calculation based on the above parameters,azide 8 should have relatively high burning rates (40 mm/s at 10 MPa).However,in reality it is capable of burning only at high pressures,and the measured rates only tend to the calculated ones. Probably,the reason for the absence of combustion of azide 8 at pressures below 12 MPa is the Landau-Derrier hydrodynamic instability [46a-c].Azide 8 has a relatively low melting point(127C),therefore,when it burns,a wide molten layer is formed,which,with a fast heat-generating reaction,can boil up intensively,completely tearing off the entire reaction layer into the gas phase.A similar phenomenon,for example,is observed during the combustion of ammonium dinitramide (ADN) [47,48].ADN does not burn at pressures below 0.2 MPa.However,the introduction of even minor additives (fine solids,some organic compounds),which suppresses intense boiling,allows the ADN to burn in a vacuum.At high pressures,the density of the outflowing gases increases and the intensity of boiling decreases with increasing pressure. Fig.11.Comparison of the kinetics of the leading combustion rate with the kinetics of its decomposition of nitroester 4. Unlike azide 8,nitrate 4 is capable of burning at a lower pressure.The temperature distribution in the combustion wave of nitrate 4 was obtained using thin tungsten-rhenium thermocouples at pressures of 0.6 and 1.1 MPa (Figure S6).The maximum measured flame temperature is 2505 K,which is lower than the calculated adiabatic temperature (3330 K).The observed overestimation of the calculated temperature over the experimental one can be explained by the losses of the thermocouple to radiation,which appear at temperatures above 2000 K [43]. For nitrate 4,the heat gradient over the combustion surface at 1.1 MPa is φ=1.25 × 10K/cm.Using the thermal conductivity coefficient λ=1.1×10J/cm×K calculated by the Real Code[44],the heat flux,q,from the gas phase to the surface is 136 J/cms,and taking into account the burning rate,the amount of heat coming from the gas phase is Q=q/m=193 cal/g.At the same time,for heating the compound to the surface temperature and melting,it is necessary to Q=c(T-T)+L=1.55(666-298)+110=680 J/g.Comparison of these values shows that the incoming heat is insufficient for heating the compound to the surface temperature and melting.However,nitrate 4 burns.Consequently,combustion propagates due to heat release in the condensed phase (in the melt),the heat flux from the gas phase is spent on the evaporation of the compound undecomposed in the melt. The measured surface temperatures at two pressures do not allow reliable determination of the pressure dependence of the surface temperature.It is also incorrect to use the initial vapor pressure in the Bourdon manometer,since the kinetic stability of nitroester 4 is not high (about an order of magnitude worse than that of azide 8).It is known that if a compound burns by a mechanism with a leading reaction in the condensed phase,the index in the combustion law is equal to the ratio of the activation energy of the leading combustion reaction to the doubled heat of vaporization n=E/2L[49a,b]. According to this equation,the heat of vaporization of nitrate 4 is L=97.9 kJ/mol (309 J/g).Using this Lvalue and the found surface temperatures (Fig.10),the dependence of the surface temperature on pressure can be described by equation (3): With the dependence of the surface temperature on pressure and thermophysical parameters in the hands,it is possible to calculate the rate constants of the leading combustion reaction using the combustion model with a leading reaction in the condensed phase [45]: As can be clearly seen from Fig.11,the obtained rate constants of the leading combustion reaction are in good agreement with the kinetics of the decomposition of nitrate 4,obtained at lower temperatures using DSC and manometry. As you know,the combustion of various nitroesters with high volatility and low boiling point is determined by the kinetics of heat-generating reactions in a flame (the so-called gas-phase combustion mechanism)[50a-c].However,a different combustion mechanism was found for nitroester 4.The reason for the change in the combustion mechanism is the higher boiling point of compound 4,which is 314C(for comparison,the boiling point of NG is 250C).With close kinetic parameters of the decomposition of nitrate 4 and NG (Fig.8),a higher boiling point leads to a greater depth of decomposition in the melt during combustion,which is~50% for 4 and 0.2% for NG at 0.6 MPa. Experimentally determined properties and Shock and detonation-derived calculated performance of nitroester 4 and azide 8 compared to NG and PETN are provided in Table 6.Thermal sensitivity has been discussed above.Although nitroester 4 is more impact sensitive than PETN,it still has lower sensitivities than NG.The sensitivities of azide 8 is slightly less than PETN.Both nitroester 4 and azide 8 show friction sensitivity comparable to PETN.In general,the sensitivity-structure relationship is consistent with previously identified patterns[51].For practical use as energetic coplasticizers,the benchmark sensitivity should not be higher than that of NG.Thus,the sensitivity of these new azofurazans is within an acceptable range.It should be noted that both nitrate 4 and azide 8 dissolve well in nitroesters and are compatible with nitrocellulose and polymers bearing nitroxy and azido groups. With a high nitrogen content,nitrate 4 and azide 8 have positive enthalpies of formation,whereas the enthalpies of formation of NG and PETN are negative.These azofurazans are characterized by a high energy output,so that in terms of the heat of explosion azide 8 is superior to PETN,and nitrate 4 surpasses to both PETN and NG. The data presented in Table 6 also demonstrate other characteristics,the values of which are in an attractive range from the point of view of propellant components.Being relatively lowmelting energetic compounds (Table 6),azide 8 and nitrate 4 may be of interest as coplasticizers of propellants and smokeless gunpowders. Table 6.Properties of compounds 4 and 8 versus NG and PETN. In conclusion,the preparation of new nitroxy-and azidomethylfunctionalized azofurazans,which may have potential use as propellant ingredients,is described.This was achieved by developing operationally simple,efficient and scalable protocols for the synthesis of target energetic compounds,4,4-bis(nitroxymethyl)azofurazan (4) and 4,4-bis(azidomethyl)azofurazan (8),using inexpensive commercially available reagents.All compounds were fully characterized by multinuclear NMR and IR spectroscopies,the X-ray single-crystal diffraction,as well as elemental analyses.The thermal decomposition and combustion of potential rocket propellant ingredients of this study were studied using a number of complementary experimental techniques.As a result,it is shown that (i) the thermal stability of compound 8 is determined by the kinetics of decomposition of azide groups,and compound 4,by nitroester groups;(ii) azide 8 is capable of burning only at a pressure above 12 MPa and the combustion rate strongly depends on the pressure,while the regularities of the combustion of nitrate 4 are typical for nitroesters;(iii)unlike nitroester 4,for azide 8,not all of the energy stored in the molecule is released in the combustion wave,which is due to the formation of endothermic nitriles in the combustion products;(iv) the combustion mechanism of both compounds is determined by reactions in the condensed phase.The above phenomena are of real significance to use. The results of this study demonstrate new horizons for the design of novel energetic compounds based on the azofurazan framework.Additionally,the intermediates synthesized during the development of approaches to target compounds 4 and 8 have great synthetic potential.Further work will look at expanding the variety of energetic azofurazans from the intermediates of this study. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. This work was supported by the Scientific Schools Development Program by Zelinsky Institute of organic chemistry(to A.B.S.,E.S.K.,R.E.N.and K.V.S.) Supplementary data to this article can be found online at https://doi.org/10.1016/j.dt.2021.08.008.
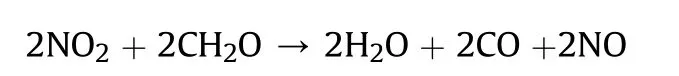

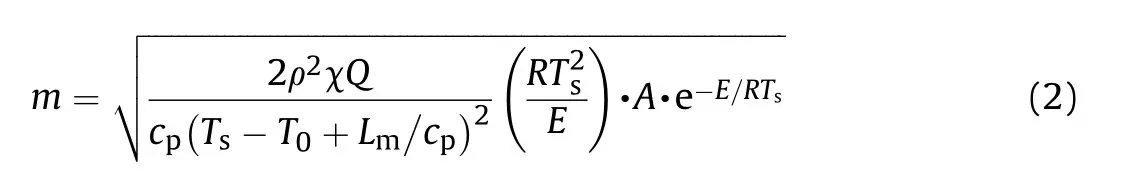

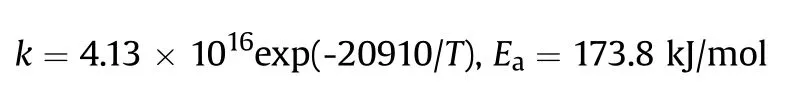
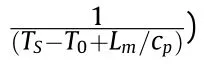
杂志排行

Defence Technology的其它文章
- Experimental study on propagation characteristics of rotating detonation wave with kerosene fuel-rich gas
- Adaptive robust control for triple avoidance -striking -arrival performance of uncertain tank mechanical systems
- Experimental study on WFeNiMo high-entropy alloy projectile penetrating semi-infinite steel target
- Numerical investigation of a muzzle multiphase flow field using two underwater launch methods
- Shock wave and bubble characteristics of underwater array explosion of charges
- A micro-chip exploding foil initiator based on printed circuit board technology