基于生物信息学构建前列腺癌单细胞层面lncRNA-miRNA-mRNA调控网络*
2022-08-29莫泳锋谢远亮陈蔓莹覃洪宇
莫泳锋,谢远亮,陈蔓莹,覃洪宇,叶 雨**
(1.广西医科大学第二附属医院,广西南宁 530007;2.广西医科大学附属肿瘤医院,广西南宁 530021)
前列腺癌(Prostate Cancer,PCa)是常见的泌尿系统恶性肿瘤之一。据统计,2018年全世界有将近130万PCa新发病例和35.9万PCa死亡病例。PCa的发病率为13.5%,死亡率为6.7%,分别居全球男性肿瘤疾病第二位和第五位[1]。随着社会的发展,人口逐渐向老龄化过渡,饮食健康问题及生活方式的改变使PCa的发病形势仍十分严峻[2,3]。因此,开展PCa的机制研究,不断深入探索肿瘤的发病机制,从分子机制上取得进一步突破,对临床诊治工作有必要且关键的作用。
生物信息学分析是基于高通量测序结果研究肿瘤发病机制和鉴定预后生物标志的重要方法。lncRNA是内源性非编码长RNA(长度200-100 000个核苷酸),转录本主要位于细胞核中,其稳定性和保守性较差,但特异性强[4]。lncRNA参与包括PCa在内的多种类型癌症的细胞增殖、迁移、侵袭和凋亡等生物学过程[5-8]。miRNA是内源性非编码微小RNA(长度约22个核苷酸),在细胞和体液(包括血浆或血清)中相对稳定[9,10]。在大多数情况下,miRNA与靶信使RNA的3′未翻译区相互作用,在转录后诱导mRNA降解和抑制翻译[11]。有研究提出了竞争性内源性RNA(Competing Endogenous RNA,ceRNA),即编码RNA和非编码RNA可以通过竞争相同的miRNA反应元件(miRNA Response Elements,MREs)来相互调节彼此的表达水平[12-14]。ceRNA的提出意味着在lncRNA-mRNA之间存在着通信调节网络,通过竞争相同的miRNA序列来调节彼此的表达,形成一个复杂多样的lncRNA-miRNA-mRNA调控网络从而调节生物学过程和肿瘤发生,如转录调控、转录后调控、蛋白修饰等[15]。近年来虽然对ceRNA的研究越来越深入并取得了一定的成果,但是lncRNA-miRNA-mRNA网络对PCa的具体调控机制还不明确。
单细胞RNA测序(Single cell RNA sequencing,scRNA-seq)是一种可以在单细胞水平上探索复杂组织细胞内转录组信息的新技术[16]。与批量RNA测序(Bulk RNA-seq)相比,scRNA-seq可以反映单个细胞的转录组表达水平,为细胞分子机制研究提供更精细且可靠的研究方法。lncRNA-miRNA-mRNA网络的调控机制在bulk RNA-seq的支持下已得到越来越多学者的认可,并证实其在肿瘤的发生发展中起着重要作用。目前,关于scRNA-seq与bulk RNA-seq技术结合构建的lncRNA-miRNA-mRNA网络研究仍较少,将二者结合构建单细胞水平的lncRNA-miRNA-mRNA调控网络可为肿瘤致病机制研究提供新方向,同时有望获得潜在的预后生物标志物,为PCa的诊断和治疗提供新思路。
1 资料与方法
1.1 数据获取
从GDC(https://portal.gdc.cancer.gov/)中获取TCGA数据库PCa的RNA-Seq测序文件,共获得499例肿瘤组织和52例正常前列腺组织。通过GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)获取GSE157703中的2例前列腺癌单细胞测序数据。
1.2 构建表达矩阵
通过R_4.1.1软件(https://www.r-project.org/),使用rtracklayer_1.50.0和SummarizedExperiment_1.20.0命令集构建人类基因信息比对数据库。用data.table_1.14.0、tidyr_1.1.3和dplyr命令集读取PCa的RNA-Seq测序文件进行整理获取全转录组表达矩阵,然后与基因信息库进行基因匹配,获取转录组的基因属性,通过基因属性分别获取lncRNA和mRNA的表达矩阵。
1.3 差异基因的筛选
用R语言的DESeq2_1.30.1和edgeR_3.32.1命令集对构建的lncRNA和mRNA表达矩阵进行整理分析,分别得到lncRNA和mRNA差异表达基因(Differentially Expressed Genes,DEGs),使用plot命令集绘制靶基因的mRNA火山图,用pheatmap_1.0.12命令集绘制Top 100-DEGs的热图。DEGs的筛选条件为|log2FC|>1且adj.P<0.05,log2FC表示取差异表达倍数以2为底的对数,adj.P表示矫正的P值。
1.4 lncRNA-miRNA-mRNA网络的构建
通过Mircode数据库(http://www.mircode.org/),运用perl软件对lncRNA-miRNA关系对进行预测。将获取的miRNA通过TargetScan数据库(https://www.targetscan.org/vert_72/)、miRTarBase数据库(https://mirtarbase.cuhk.edu.cn/)和miRDB数据库(http://mirdb.org/)分别预测miRNA的靶基因,取交集得到最终的miRNA靶基因,然后与整合的DEGs取交集,最终得到多条lncRNA-miRNA-mRNA轴,据此构建一个lncRNA-miRNA-mRNA调控网络,并通过Cytoscape_3.8.1进行可视化。
1.5 功能富集分析
对构建的lncRNA-miRNA-mRNA网络中的靶基因进行生物学功能及通路富集分析。首先对lncRNA-miRNA-mRNA网络中的mRNA运用R语言的org.Hs.eg.db_3.12.0包进行ID转换;然后通过clusterProfiler_3.18.1、enrichplot_1.10.2和ggplot2_3.3.3包进行基因本体论(Gene Ontology,GO)以及京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)分析,以PvalueCutoff=0.05、QvalueCutoff=0.05为筛选条件,结果以气泡图和圈图显示;最后使用STRING (https://string-db.org/)数据库对网络中的mRNA进行蛋白互作分析,设定Medium confidence>0.4,结果使用Cytoscape_3.8.1将数据可视化。
1.6 单细胞测序数据的整合以及构建单细胞水平调控网络
对获取的单细胞测序文件,首先通过R语言的Seurat_4.0.1、ggplot2_3.3.3、clustree_0.4.3和cowplot_1.1.1包等命令集,合并整理构建单细胞表达矩阵、矫正批次效应以及质量控制;然后进行降维,根据Top 2 000差异基因进行聚类和分群;最后根据mark基因对各细胞亚群进行定义,通过拷贝数变异(Copy Number Variation,CNV)定义肿瘤细胞亚群,并进一步验证lncRNA-miRNA-mRNA网络中的靶基因在各细胞亚群上的表达情况,同时构建单细胞水平的lncRNA-miRNA-mRNA调控网络。
1.7 预后分析与免疫组织化学验证
利用R软件的cgdsr、DT、survival等数据包,获取494例PCa的临床数据及转录组数据,根据基因的中位表达情况,将其分为目的基因高表达组和低表达组,并分析无病生存期(Disease-Free Survival,DFS),根据疾病有无复发或死亡将其分为复发组和死亡组或无复发组和死亡组,并分析基因的表达情况,通过ggpubr包进行可视化。然后将基因输入在线数据库人类蛋白质图谱(https://www.proteinatlas.org/),参数选择前列腺癌病理图谱,获取目的基因免疫组织化学的镜下图像并导出,通过图像处理软件Adobe Illustrator可视化。
2 结果与分析
2.1 差异表达基因的筛选与鉴定
通过获取TCGA的PCa测序数据,经过筛选得到3 013个DEmRNA,其中上调1 282个,下调1 731个[图1(a)]。由图1(b)可知,共得到1 101个DElncRNA,其中上调443个,下调658个。取前100个差异基因作热图分析,结果显示差异基因在肿瘤和正常组织中存在差异表达(图2)。
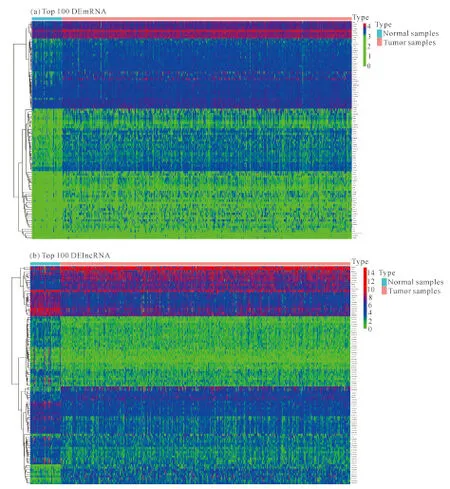
图2 差异基因热图分析结果Fig.2 Heat map analysis results of differential gene
2.2 lncRNA-miRNA-mRNA网络的构建与分析
将得到的DElncRNA通过Mircode数据库进行miRNA预测,得到1 935个lncRNA-miRNA关系对,然后将miRNA通过TargetScan、miRTarBase和miRDB数据库预测mRNA,与mRNA DEGs取交集得到97个miRNA的靶基因,最终纳入构建调控网络的基因共175个,包括51个lncRNA、27个miRNA和97个mRNA。结果通过调控网络图进行可视化(图3)。
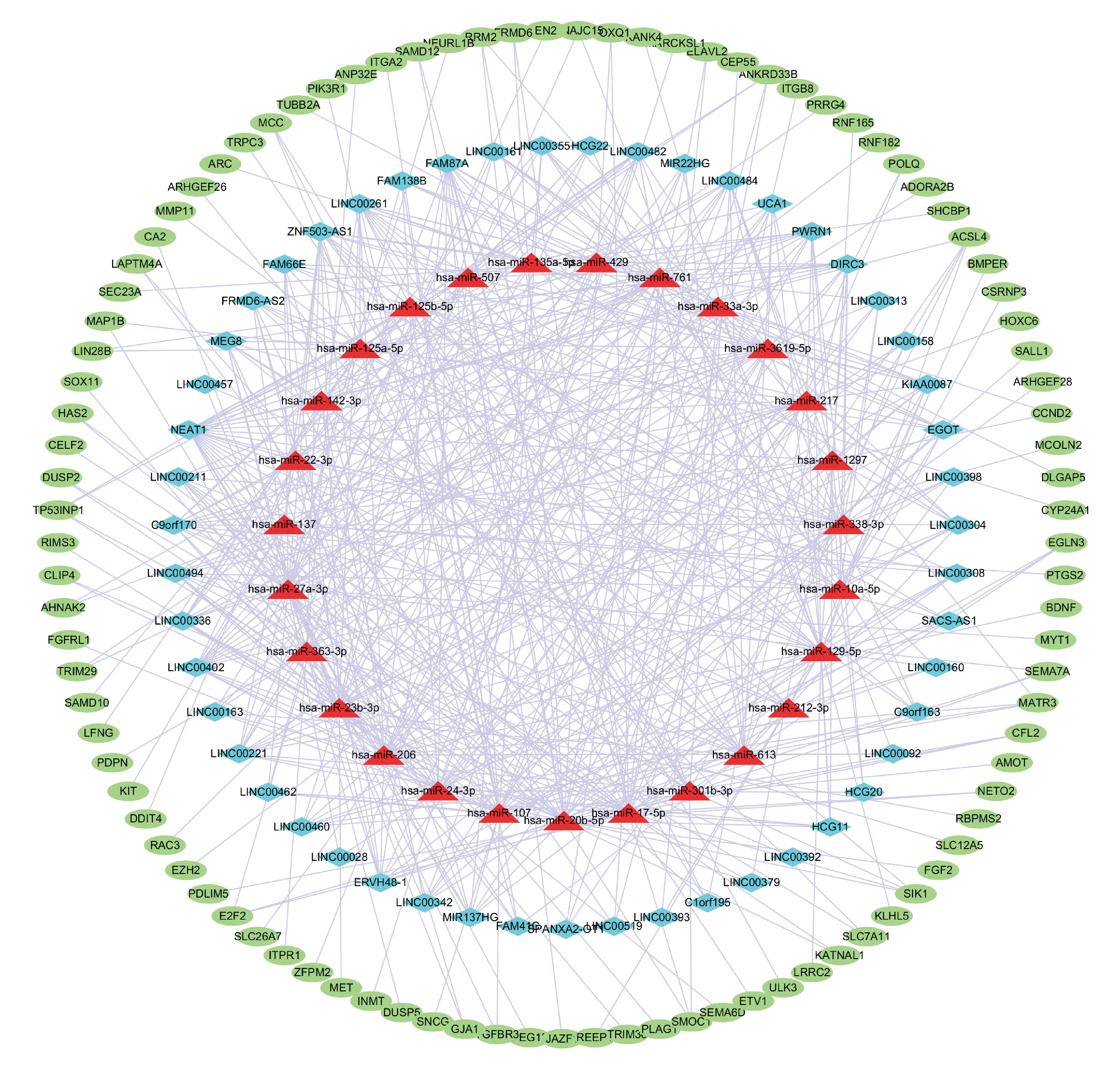
Blue diamond represents lncRNA,red triangle represents miRNA,green oval represents mRNA,and the linear connection represents that they have regulatory relationships
2.3 GO生物功能富集与KEGG信号通路富集分析
根据GO分析和KEGG分析,以P<0.05,FDR<0.05为筛选条件,并按照富集基因个数从大到小排列,将排名在前15的富集结果和KEGG分析结果用气泡图和圈图表示(图4)。GO分析结果显示,lncRNA-miRNA-mRNA调控网络的靶基因在腺状细胞迁移、化学突触传递的调节、跨突触信号的调节、细胞形态分化与发生的调节、上皮细胞迁移和组织迁移等生物学过程富集[图4:(a)(c)(e)]。KEGG分析结果显示,miRNA在癌症、轴突导向、胞间的黏附、肌动蛋白细胞骨架调节、Rap1、MAPK、PI3K-Akt和Ras等信号通路富集[图4:(b)(d)(f)]。
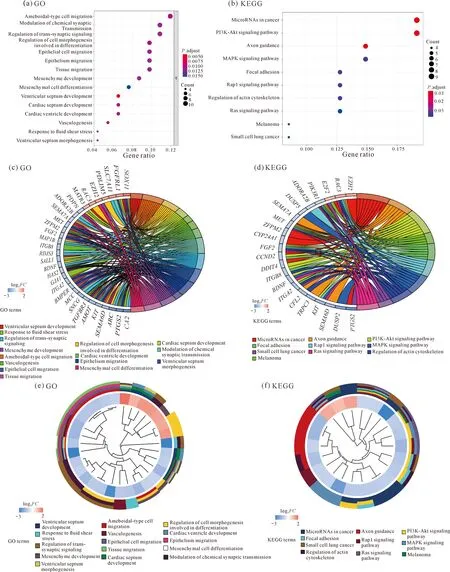
图4 GO和KEGG分析结果Fig.4 Analysis results of GO and KEGG
2.4 蛋白质互作网络的构建与分析
使用STRING数据库分析lncRNA-miRNA-mRNA调控网络靶基因的蛋白互作关系,通过Cytoscape软件绘制,得到蛋白-蛋白互作(PPI)网络图,圆圈越大则互作对越多。由图5可知,共得到58个蛋白质的互作网络,其中Top 10基因分别为BDNF、FGF2、EZH2、MET、KIT、PIK3R1、PTGS2、RRM2、DLGAP5和CEP55。
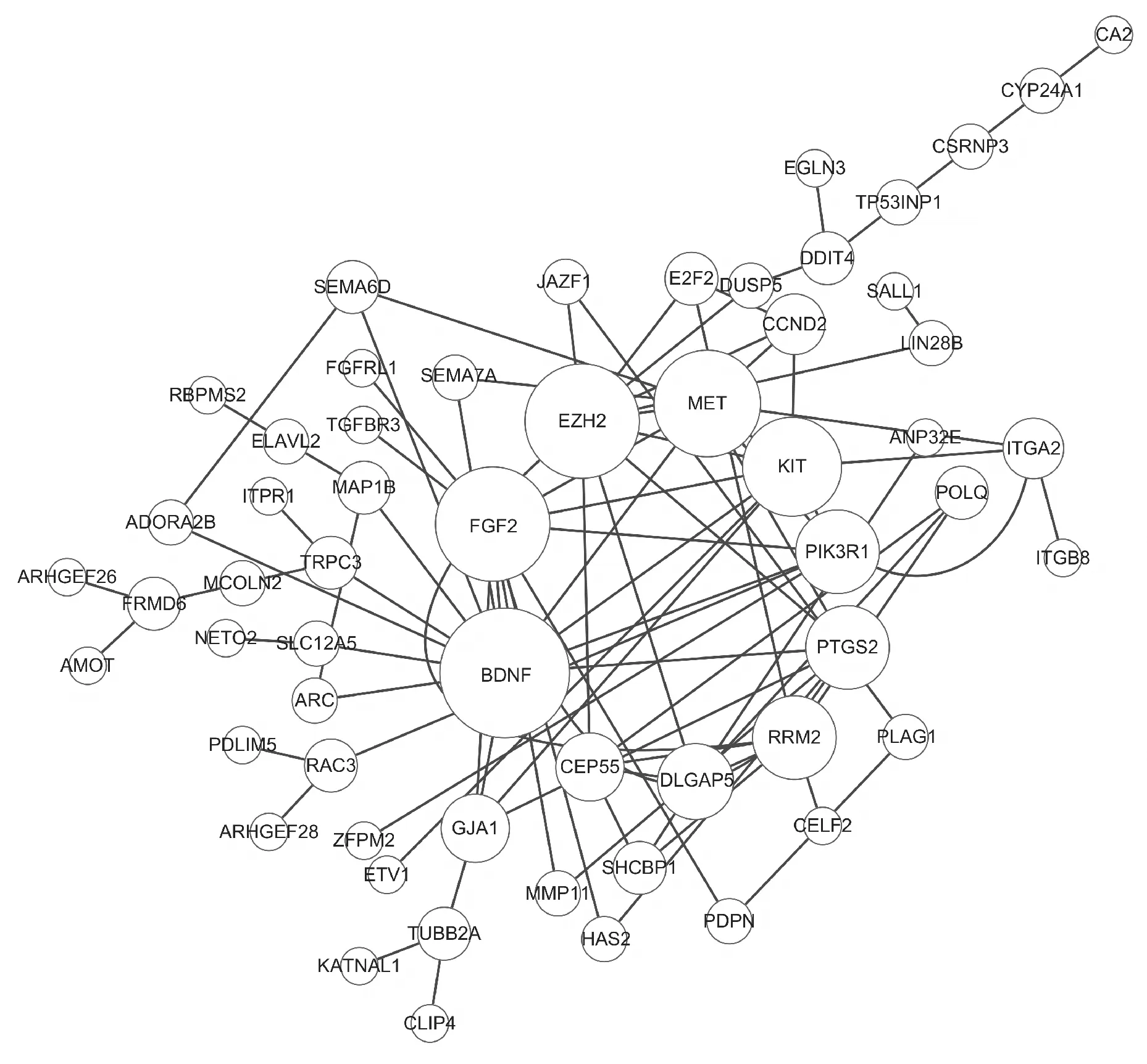
Circle represents proteins,the larger the circle,the higher the interaction score;and the linear connection represents the interaction between proteins
2.5 单细胞测序数据的整合及构建单细胞水平调控网络
使用R软件将获取的GSE157703中的2例前列腺癌单细胞测序数据进行整合分析,得到6 803个细胞、24 639个基因的单细胞矩阵,最终定义了B细胞、T细胞、成纤维细胞、肥大细胞、内皮细胞、上皮细胞、平滑肌细胞、髓系细胞和肿瘤细胞共9个细胞亚群(图6)。
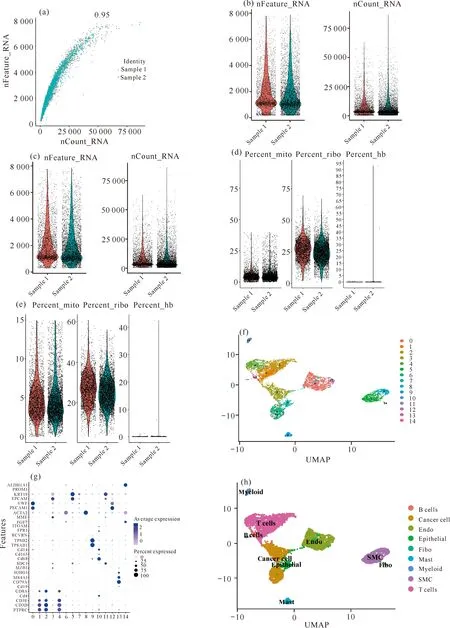
Sample 1 and Sample 2 are sequenced samples of cancer tissue from two different PCa patients
为验证lncRNA-miRNA-mRNA调控网络靶基因在各个亚群的表达情况,取平均表达10%以上的亚群作为单细胞水平调控网络构建的标准[图7:(a)(b)],据此可以得到B细胞表达靶基因PEG10,调控网络中有25个lncRNA、1个miRNA和1个mRNA,共25条lncRNA-miRNA-mRNA轴[图7(c)];得到T细胞表达靶基因DUSP2,调控网络中有9个lncRNA、2个miRNA和1个mRNA,共18条lncRNA-miRNA-mRNA轴[图7(d)];得到成纤维细胞表达靶基因PDPN、HAS2、MAP1B和INMT,调控网络中有32个lncRNA、5个miRNA和4个mRNA,共70条lncRNA-miRNA-mRNA轴[图7(e)];得到肥大细胞表达靶基因KIT和PTGS2,调控网络中有6个lncRNA、1个miRNA和2个mRNA,共12条lncRNA-miRNA-mRNA轴[图7(f)];得到内皮细胞表达靶基因GJA1、SNCG、TGFBR3、DUSP5、INMT和MET,调控网络中有46个lncRNA、6个miRNA和6个mRNA,共158条lncRNA-miRNA-mRNA轴[图7(g)];得到平滑肌细胞表达靶基因RBPMS2、SNCG、MAP1B和ARC,调控网络中有39个lncRNA、5个miRNA和4个mRNA,共77条lncRNA-miRNA-mRNA轴[图7(h)];得到上皮细胞表达靶基因EGLN3,调控网络中有11个lncRNA、3个miRNA和1个mRNA,共23条lncRNA-miRNA-mRNA轴[图7(i)];得到髓系细胞表达靶基因SLC7A11和DUSP5,调控网络中有31个lncRNA、3个miRNA和2个mRNA,共43条lncRNA-miRNA-mRNA轴[图7(j)];得到肿瘤细胞表达靶基因TRIM36、TRIM29、TP53INP1、ITGA2、PDLIM5、AMOT、PRRG4和MET等,调控网络中有45个lncRNA、10个miRNA和8个mRNA,共188条lncRNA-miRNA-mRNA轴[图7(k)]。

图7 lncRNA-miRNA-mRNA调控网络在单细胞亚群中的表达情况Fig.7 Expression of lncRNA-miRNA-mRNA regulatory network in single cell subpopulation
2.6 PCa肿瘤细胞相关靶基因预后分析及验证
通过R软件分别获取ITGA2、MET、PDLIM5、PRRG4以及TP53INP1基因的DFS及其分组表达情况(图8)。结果显示,ITGA2、PDLIM5H和PRRG4的DFS存在差异且均具有统计学意义(P<0.05),上述基因的低表达组有更高的肿瘤复发率或死亡率。为了进一步验证调控网络的靶基因在PCa单细胞水平上的肿瘤细胞群的表达情况,通过在线数据库分别验证ITGA2、PDLIM5和PRRG4基因在肿瘤细胞中的蛋白水平表达情况,结果显示上述靶基因在PCa患者的肿瘤组织病理中均有不同程度的表达。
3 讨论
随着测序技术的不断发展,研究重点逐步从编码RNA向非编码RNA转变。随着近年来对肿瘤lncRNA-miRNA-mRNA轴的不断深入研究,越来越多的研究表明lncRNA-miRNA-mRNA与PCa的发生和发展密切相关。体外实验发现,lncRNA KCNQ1OT1/miR-211-5p/CHI3L1调控PCa细胞的增殖、迁移、侵袭[17]。Luo等[18]的研究发现,lncRNA TTN-AS1/miR-193a-5p在PCa中起到抑制细胞凋亡的作用。Jiang等[19]的研究发现,lncRNA HOXD-AS1/miR-361-5p/FOXM1参与PCa肿瘤的转移。也有研究提出lncRNA HOTAIR通过竞争性吸附miR-590-5p,参与调节STAT3,并参与PCa细胞对多西他赛的耐药[20]。上述研究结果表明,lncRNA-miRNA-mRNA调控网络参与了调节PCa的增殖、迁移、侵袭和凋亡等多个生物学过程以及化疗药物的抵抗。以往构建的lncRNA-miRNA-mRNA调控网络是基于bulk测序来进行的,具有宏观的指导意义,但更精确的微观调控关系仍待进一步研究。单细胞测序技术的提出,使测序结果能精确到单个细胞水平,为肿瘤分子机制的研究提供了新方法。本研究通过对TCGA-PRAD的bulk测序和GEO中GSE157703的scRNA数据进行整合分析,以此构建单细胞水平的lncRNA-miRNA-mRNA调控网络,这些调控网络可以为PCa的分子机制研究提供理论依据。
本研究通过对TCGA测序数据进行处理,得到3 013个DEmRNA和1 101个DElncRNA,筛选出51个lncRNA、27个miRNA和97个mRNA来构建lncRNA-miRNA-mRNA调控网络。通过GO分析发现,调控网络的靶基因在腺状细胞的迁移、细胞形态分化与发生调节、上皮细胞迁移和组织迁移等生物学过程富集。KEGG分析发现,调控网络的靶基因在胞间黏附、肌动蛋白细胞骨架调节等信号通路富集。因此可以得出生物功能主要与细胞形态分化、胞间连接和细胞迁移有关,信号通路参与了细胞黏附、细胞连接的形成。同样地,在PPI的Top 10基因中BDNF、FGF2、MET和KIT也参与了上述生物功能以及相关信号通路。远处转移是PCa患者死亡的重要原因之一,研究PCa转移的机制对于改善其预后至关重要[21]。研究发现lncRNA HOXD-AS1通过miR-361-5p/FOXM1参与PCa患者的转移[19]。本研究结果提示lncRNA-miRNA-mRNA网络参与了PCa的细胞连接、迁移以及黏附等相关生物学功能和通路的调控,可见其在转移相关分子机制的调控中有重要作用。
根据标记基因,在单细胞测序的聚类定义后,可得到B细胞、T细胞、成纤维细胞、肥大细胞、内皮细胞、上皮细胞、平滑肌细胞、髓系细胞和肿瘤细胞共9个细胞亚群的lncRNA-miRNA-mRNA调控关系。下面将其分为免疫细胞(B细胞、T细胞和髓系细胞)、上皮细胞、间质细胞(成纤维细胞、平滑肌细胞、肥大细胞和内皮细胞)以及肿瘤细胞4大类进行讨论。
免疫细胞的lncRNA-miRNA-mRNA调控网络主要通过调控PEG10、SLC7A11、DUSP2和DUSP5基因参与调节肿瘤的免疫微环境。有研究发现,lncRNA OIP5-AS1通过miR-128-3p/SLC7A11信号传导抑制长期接触镉的前列腺癌铁凋亡[22]。本研究发现,SLC7A11在髓系细胞群表达较高,提示lncRNA-miRNA-SLC7A11信号轴可通过调控PCa肿瘤微环境中的髓系细胞参与调控PCa细胞凋亡。
上皮细胞的lncRNA-miRNA-mRNA调控网络主要通过调控EGLN3基因参与肿瘤相关微环境的调节。miR-1205可以通过靶向EGLN3的3′-UTR位点,下调其转录活性参与调控PCa细胞增殖[23]。本研究发现,EGLN3主要在上皮细胞中表达,提示lncRNA-miRNA-EGLN3可能参与PCA肿瘤微环境中的上皮细胞群的相关增殖。
间质细胞的lncRNA-miRNA-mRNA调控网络主要通过调控GJA1、SNCG、TGFBR3、DUSP5、INMT、MET、KIT、PTGS2、PDPN、HAS2、MAP1B、INMT、RBPMS2、SNCG、MAP1B和ARC基因参与肿瘤相关微环境的调节。研究发现,上调的lncRNA DLX6-AS1通过miR-497-5p/SNCG轴在PCa中起到促进细胞增殖、抑制凋亡从而调控PCa进展的作用[24]。相关研究表明,在细胞实验中MiR-124-3p通过直接吸附PTGS2抑制AKT/NF-κB通路,从而抑制PCa细胞的细胞活力、增殖、迁移、侵袭并促进凋亡[25]。这些结果提示本研究构建的lncRNA-miRNA-mRNA调控网络,主要通过调控PCa的间质细胞群从而间接促进肿瘤细胞的迁移和侵袭等过程。
肿瘤细胞的lncRNA-miRNA-mRNA调控网络主要参与TRIM36、TRIM29、TP53INP1、ITGA2、PDLIM5、AMOT、PRRG4和MET基因的调控。研究发现,lncRNA625可通过靶向miR-432调控TRIM29,从而调节Wnt/β-连环蛋白通路,抑制PCa细胞增殖,促进凋亡[26]。另外,lncRNA AGAP2-AS1/miR-195-5p/PDLIM5在PCa细胞增殖、迁移和侵袭以及肿瘤生长过程发挥调控作用[27]。通过预后分析得出肿瘤细胞lncRNA-miRNA-mRNA调控网络中的靶基因ITGA2、PDLIM5H和PRRG4可以预测PCa的DFS,这些基因在蛋白水平也得到表达验证,提示其可作为预后的观测指标之一。
4 结论
本研究成功构建了在PCa单细胞水平下包括肿瘤细胞在内的9个细胞亚群的数百条精确的lncRNA-miRNA-mRNA调控网络,并发现该调控网络不仅参与PCa的发生发展调控,同时在临床预后预测方面也有重要的意义。本研究构建的多条lncRNA-miRNA-mRNA调控轴虽然为PCa的基础研究提供了新方向,但是目前在各个细胞亚群的具体生物学功能尚未得知,还需通过体外实验进一步证实。
