不对称苄基紫精与葫芦[8]脲包结行为的探究
2022-08-29张锦锦王巧纯
张锦锦, 邹 雷, 王巧纯
(华东理工大学化学与分子工程学院,精细化工研究所,结构可控先进功能材料及其制备教育部重点实验室,上海 200237)
葫芦脲(CB[n],其中n表示甘脲单元数)是由甘脲和甲醛在酸性条件下缩合形成的大环分子,其中甘脲单元之间通过亚甲基桥联在一起,两端为亲水性的羰基,中间为疏水性的空腔[1-5]。葫芦脲对带正电荷的客体分子如质子化的有机胺[6]、吡啶鎓[7]、紫精[8]等具有很强的包结能力[9]。这些客体中,紫精分子具有可逆的氧化还原变色性能,而且葫芦脲的包结对紫精还原态自由基有良好的稳定性作用[10],因此紫精/葫芦脲复合体系在刺激响应型材料[11]、超分子自组装[12]等领域的研究备受瞩目。许多研究结果表明,紫精分子的取代基以及葫芦脲空腔的大小对两者的包结模式有很大的影响。比如,内腔较小的CB[6]一般与烷基化取代的紫精结合力较强,且CB[6]停留在烷基上[13]。内腔中等的CB[7]与紫精化合物的包结位点则取决于取代基的极性及烷基链的长度,当取代基的极性大,或者体积较小时,CB[7]会停留在联吡啶部分[14];当取代基(特别是苄基[15])为亲油性基团且体积较大时,CB[7]则停留在侧链基团上。腔体较大的CB[8]则倾向于包结两个芳香型取代基单元,特别是当紫精单元与芳香型电子给体(如萘酚衍生物)相连时,CB[8]内腔同时包结紫精和电子给体,形成三元复合物,甚至是超分子聚合物[16]。
考虑到CB[8]内腔大到足够容纳两个芳环单元,因此猜测其与含苄基的紫精分子进行包结时,很有可能形成1 个CB[8]同时包裹2 个苄基的头对头结合型复合物。这种1∶2 的包结模式如果成立,那么含多个苄基紫精的客体砌块单元就可通过与CB[8]的包结作用相互交联,这将为进一步构建相关超分子聚合物,甚至是刺激响应型材料打下良好的基础。本文制备了不对称的紫精化合物1-乙基-1’-苄基-4,4’-联吡啶溴化盐(EBV),运用核磁共振氢谱(1H-NMR)、等温滴定量热法(ITC)、高分辨质谱(HRMS)等手段,详细考察EBV 与CB[8]的包结行为。
1 实验部分
1.1 原料和试剂
联吡啶、溴化苄,均为分析纯,购于阿拉丁生化科技股份有限公司;溴乙烷、尿素、多聚甲醛,均为分析纯,购于上海麦克林生化科技有限公司;乙腈、N,N-二甲基甲酰胺(DMF)、浓盐酸(w=37%)、乙酸乙酯、无水甲醇(w=98%)、丙酮,均为分析纯,购于上海泰坦科技股份有限公司。以上试剂未经进一步提纯直接使用。实验用水为去离子水。
1.2 测试与表征
1H-NMR 与核磁共振碳谱(12C-NMR)使用德国Brüker 公司 AM-400 型核磁共振波谱仪测试,以四甲基硅烷(TMS)为内标,室温下测试;HRMS 采用Xevo G2 TOF MS 的电喷雾离子化(ESI)-高分辨飞行时间质谱仪测试;等温滴定量热仪的型号为Microcal ITC200(GE Healthcare)。
1.3 实验步骤
1.3.1 EBV 的合成 EBV 的合成在文献报道[17-18]的基础上进行改进,如图1 所示。
称取联吡啶(9.37 g,60.0 mmol)溶于50.0 mL 乙腈中,加热至回流。缓慢滴加溴乙烷(1.09 g,10.0 mmol)与10.0 mL 乙腈的混合溶液,滴加完毕后继续回流反应20 h。冷却后旋蒸除去溶剂,剩余物用乙酸乙酯多次洗涤除去未反应联吡啶,干燥后得1.85 g 浅黄色固体1(产率69.8%)。1H-NMR (400 MHz, D2O,298 K,δ):8.84 (d,J=6.9 Hz,2H),8.62 (d,J=6.3 Hz,2H),8.25(d,J=6.7 Hz,2H),7.76 (d,J=6.3 Hz,2H),4.56 (q,J=7.4 Hz,2H),1.54 (t,J=7.4 Hz,3H)。
将固体1(0.251 g,1.0 mmol)和溴化苄(1.71 g,10.0 mmol)加入到耐压管中,用乙腈或DMF 作溶剂,油浴加热反应24 h。冷却后抽滤,用乙腈多次洗涤滤饼,干燥得0.404 g 黄绿色固体EBV(产率92.6%)。1H-NMR (400 MHz,D2O,298 K,δ):9.12 (dd,J=14.2,6.9 Hz,4H),8.51 (t,J=5.3 Hz,4H),7.51 (s, 5H),5.91 (s,2H),4.74 (q,J=7.4 Hz,2H),1.67 (t,J=7.3 Hz,3H);13CNMR(100 MHz,D2O,298 K,δ):150.46,150.39,149.74,145.42,145.15,132.14,130.08,129.59,129.26,126.98,64.72,57.60,15.57;HRMS(ESI):m/z355.081 3([MBr]2+计算值355.081 0)。1.3.2 CB[8]的合成及纯化 CB[8]按照文献[1-5, 19]中所述的方法进行制备及纯化。
2 结果与讨论
2.1 EBV 与CB[8]包结行为的核磁研究
虽然已有文献[20-21]提及CB[8]与EBV 形成1∶1(物质的量之比)的包结物,但在对二者进行核磁滴定实验时发现,随着CB[8]加入量的增加,除了EBV 本身的质子信号外,还出现了一组新的质子信号(图2(b)及2(c))。进一步对这一组信号进行峰面积积分时发现,CB[8]与EBV 单元的物质的量之比保持为1∶2。相比纯粹的EBV 核磁,这组新的核磁信号中EBV 联吡啶单元的Hd、He的化学位移向低场分别移动了0.27 和0.17;Hf则向高场移动0.36;而苯环单元上的Hh、Hi、Hj进一步发生分化,分别向高场移动1.31、1.16、1.16;此外,苄基上亚甲基Hg的化学位移也向高场移动0.72,而乙基单元上的氢及联吡啶单元上的Hc的化学位移基本上没有变化。这说明CB[8]与EBV 极有可能形成了我们预期的1∶2 头对头包结物2EBV-CB[8](图1),即1 个CB[8]内腔同时包结2 个苄基单元。
此时,苄基及处于CB[8]端口的Hf因为屏蔽效应向高场移动;处于端口外面的Hd、He由于去屏蔽效应向低场移动;而远离CB[8]的Hc及乙基上的氢则基本不受影响。当CB[8]含量进一步增加,CB[8]与EBV 的物质的量之比为1∶2 时,游离的EBV 质子信号完全消失(图2(d)),只剩下包结物的信号峰。再继续加入CB[8]时,所得核磁图谱并不发生变化,说明二者确实形成1∶2 的稳定包结物。而且这时由于CB[8]在水中的溶解度差(c<1.00×10-5mol/L)[4],过量的CB[8]离心后沉降在核磁管底部。进一步将CB[8]与EBV 物质的量之比为1∶2 条件下的核磁与文献[20-21]报道的图谱进行比对,发现二者完全一致。这说明文献报道的包结模式中,CB[8]停留在苄基上是正确的,但是二者最终并非形成1∶1 的包结物。
2.2 EBV 与CB[8]的包结行为的ITC 分析
为进一步研究EBV 与CB[8]的包结行为,进行了ITC 测 试。将1.30×10-3mol/L 的EBV 水 溶 液 在298.15 K 下逐渐滴加到1.00×10-4mol/L的CB[8]水溶液中,结果如图3 所示,得到的具体数据如表1 所示。所得结果并不能按经典的用于拟合1∶1 包结模式的“Independent”模型进行拟合,而与“Multiple sites”等温模型则匹配良好。这表明EBV与CB[8]的包结过程中,两个客体分子不是同时与主体分子包结[21],而是分步进行,即如图1(b)所示:随着EBV 加入到CB[8]溶液中,首先进行1∶1 的包结,该过程的结合常数为K1=(1.65±1.22)×107M-1(1 M=1 mol/L,余同),对应的ΔH1和-TΔS1分别为(-26.2±1.26) kJ/mol和14.6 kJ/mol。之后随着EBV 的进一步加入,最终形成1∶2 的稳定包结物,整个过程的表观包结常数(Ka)为(1.34±0.193)×1013M-2,对应的ΔH和-TΔS分别为(-64.4±3.19)kJ/mol 和-9.43 kJ/mol,这表明该组装行为是焓和熵共同驱动的。
2.3 EBV 与CB[8]的包结行为的质谱分析
上述包结行为的推论进一步得到了ESI-HRMS的验证。如图4 所示,在ESI-HRMS 谱图中发现了1∶1 包 结 物 的[M-2Br]2+信 号 峰,m/z值 为802.28,相关同位素峰型与理论模拟结果一致。此外,质谱图中在m/z=1 019.28 处出现了1∶2 包结物的[M-2Br]2+信号峰,同样其相关同位素峰型与理论模拟结果一致。这表明EBV 与CB[8]包结过程中确实经历了从1∶1 包结到1∶2 包结这两种过程。
2.4 浓度对1∶1 及1∶2 两种包结模式的影响

EBV 与CB[8]的包结比例受浓度影响。EBV与CB[8]的反应过程为:其中:H 表示主体分子CB[8];G 表示客体分子EBV;HG 表示CB[8]与EBV 以1∶1(物质的量之比)形成的超分子;HG2表示GB[8]与EBV 以1∶2(物质的量之比)形成的超分子。
则反应平衡时,

其中:c(H)、c(G)、c(HG)、c(HG2)分别表示H、G、HG、HG2的平衡浓度。
另外,由物料守恒得知,

其中:c(H0)、c(G0)分别表示CB[8]和EBV 的总浓度。
因最初将CB[8]和EBV 以物质的量之比1∶2混合,所以:

联立式(3)~(7)可得:

在核磁测试中,取c(HG2)为1.00×10-2mol/L,由式(4)、(8)可得:

其中:K1用1.65×107M-1代入,K2用8.14×105M-1代入,由式(9)可计算得到c(G)=3.51×10-4mol/L ,c(H)=6.06×10-8mol/L,1∶1 包结物的平衡浓度c(HG)=3.50×10-4mol/L,占包结物总摩尔分数的3.38%,而1∶2 包结物的平衡浓度c(HG2)为1.00×10-2mol/L,其摩尔分数高达96.6%。所以在核磁图谱中只观察到1∶2 的包结物。
然而,在质谱图谱中同时观察到1∶1 和1∶2 两种包结物的信号峰(图4),原因如下:取c(HG)为1.00×10-5mol/L,根据式(3)~(8)计算得到c(G)为1.01×10-5mol/L,c(H)为5.99×10-8mol/L,1∶1 包结物的平衡浓度c(HG)为1.00×10-5mol/L,占包结物总摩尔分数的54.8%,而1∶2 包结物的平衡浓度c(HG2)为8.24×10-6mol/L,摩尔分数为45.2%,所以在ESIHRMS 图谱中同时观察到1∶1 和1∶2 两种包结物的信号峰。
由此可见,EBV 与CB[8]的包结比例受其浓度影响,随着EBV 和CB[8]浓度的降低,1∶2 包结物的摩尔分数会逐渐降低,而1∶1 包结物的摩尔分数则会逐渐增加。
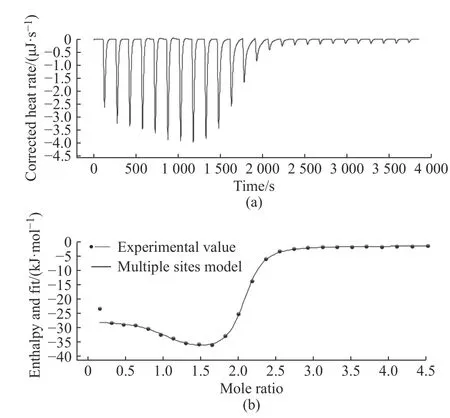
图3 将EBV(1.30×10-3 mol/L)在298.15 K 下滴加到CB[8](1.00×10-4 mol/L)水溶液中的ITC 图像Fig. 3 ITC pattern of the complexation of EBV(1.30×10-3 mol/L) with CB[8](1.00×10-4 mol/L) in aqueous solution at 298.15 K

表1 EBV 和CB[8]包结行为的ITC 测定值(298.15 K)Table 1 ITC values of the complexation of EBV with CB[8] (298.15 K)
3 结 论
通过1H-NMR、ITC 和HRMS 分析表明,EBV 与CB[8]在水溶液中进行分步结合,先经过1∶1 包结,最终实现1∶2 包结。第一过程包结常数为(1.65±1.22)×107M-1,ΔH1和-TΔS1分别为(-26.2±1.26) kJ/mol和14.6 kJ/mol。而整个过程的表观包结常数为(1.34±0.193)×1013M-2,对应的ΔH和-TΔS分别为(-64.4±3.19) kJ/mol 和-9.43 kJ/mol,是由焓和熵共同驱动的自组装过程。该研究结果为将来利用多苄基紫精衍生物与CB[8]进行自组装构建功能性超分子聚合物体系打下了良好基础。

图4 EBV 与CB[8]包结物的ESI-HRMS((a)、(b)分别为1∶1、1∶2 包结物的信号峰,其中上面为理论模拟图,下面为测试图)
Fig. 4 ESI-HRMS of the EBV/CB[8] complex ( (a) and (b) are the signal peaks of 1∶1 and 1∶2 inclusions, respectively, the corresponding simulated spectra (up) and test spectra (down))
[11]ZHANG Q, QU D H, WANG Q C,et al. Dual-mode controlled self-assembly of TiO2nanoparticles through a cucurbit[8]uril-enhanced radical cation dimerization interaction[J]. Angewandte Chemie International Edition,2015, 54(52): 15789-15793.
[12]ZHANG C C, LIU X L, LIU Y P,et al. Two-dimensional supramolecular nanoarchitectures of polypseudorotaxanes based on cucurbit[8]uril for highly efficient electrochemical nitrogen reduction[J]. Chemistry of Materials, 2020,32(19): 8724-8732.
[13]KIM K. Mechanically interlocked molecules incorporating cucurbituril and their supramolecular assemblies[J]. Chemical Society Reviews, 2002, 31(2): 96-107.
[14]MOON K, KAIFER A E. Modes of binding interaction between viologen guests and the cucurbit[7]uril host[J].Organic Letters, 2004, 6(2): 185-188.
[15]SINDELAR V, MOON K, KAIFER A E. Binding selectivity of cucurbit[7]uril: Bis(pyridinium)-1,4-xylylene versus 4,4 ’-Bipyridinium guest sites[J]. Organic Letters, 2004,6(16): 2665-2668.
[16]LUTZ F, NEREA L P, SCHMIDT T C,et al. Heteroternary cucurbit[8]uril complexes as supramolecular scaffolds for self-assembled bifunctional photoredoxcatalysts[J].Chemical Communications, 2021, 57(23): 2887-2890.
[17]HEINEN S, WALDER L. Generation-dependent intramolecular CT complexation in a dendrimer electron sponge consisting of a viologen skeleton[J]. Angewandte Chemie International Edition, 2000, 39(4): 806-809.
[18]NAGARJUNA G, HUI J, CHENG K J,et al. Impact of redox-active polymer molecular weight on the electrochemical properties and transport across porous separators in nonaqueous solvents[J]. Journal of the American Chemical Society, 2014, 136(46): 16309-16316.
[19]BARDELANG D, UDACHIN K A, LEEK D M,et al.Cucurbit[n]urils (n=5—8): A comprehensive solid state study[J]. Crystal Growth & Design, 2011, 11(12):5598-614.
[20]JI H L, LIU F Y, SUN S G. Study of the counter anions in the host-guest chemistry of cucurbit[8]uril and 1-ethyl-1’-benzyl-4,4’-bipyridinium[J]. The Scientific World Journal,2013: 452056.
[21]ZHANG T Y, SUN S G, LIU F Y,et al. Interaction of DNA and a series of aromatic donor-viologen acceptor molecules with and without the presence of CB[8][J]. Physical Chemistry Chemical Physics, 2011, 13(20): 9789-9795.
