男性21羟化酶缺乏症一例
2022-08-27李亚玲袁刚谢君辉
李亚玲 袁刚 谢君辉
患者,男,26岁,因“发现无精症4月余”遂于2018年10月26日入院。患者婚后一直未育,4个月前就诊于我院泌尿外科确诊为“无精症”,遂于我科进一步诊治。既往史:患者出生时,外生殖器发育正常,8岁时出现第二性征发育,身高明显高于同龄人。成年后身高低于同龄人。家族史:家族成员发育正常,无不孕不育相关疾病史。体格检查:身高156 cm,体重67.5 kg,BMI 27.74 kg/m2,上部量75 cm,下部量81 cm。全身体毛分布正常,全身皮肤黏膜无色素沉着。左右睾丸长径分别为2.6 cm、2.5 cm,阴毛少许,分布于阴茎周围,阴茎长约5 cm,TannerⅢ期。实验室检查:血电解质、甲状腺功能、皮质醇(8AM、4PM、12MN)均无异常。促肾上腺皮质激素(ACTH)34.91 pmol/L(1.60~13.90 pmol/L,括号内为正常参考值范围,以下相同)。性激素全套:睾酮6.59 ng/ml(1.75~7.81 ng/ml),泌乳素11.05 ng/ml(2.64~13.13 ng/ml),雌二醇62 pg/ml(≤47 pg/ml),促黄体激素<0.20 mIU/ml(1.24~8.62 mIU/ml),促卵泡激素0.13 mIU/ml(1.27~19.26 mIU/ml),孕酮16.25 ng/ml(0.14~2.06 ng/ml),雄烯二酮>35.0 nmol/L(3.7±0.9 nmol/L),硫酸去氢表雄酮21 600 nmol/L(5 735±2 380 nmol/L),17羟孕酮>60.0 nmol/L(3.5±1.2 nmol/L)。内胰岛素生长因子1(IGF-1)127.0 ng/ml(434.0±84.0 ng/ml);胰岛素样生长因子结合蛋白3(IGFBP3)正常。尿17-羟皮质类固醇激素51.52 μmol/24 h(8.30~27.70 μmol/24 h);尿17-酮皮质类固醇激素478.53 μmol/24 h(35.00~87.00 μmol/24 h);尿香草扁桃酸(VMA)、尿高香草酸、尿肾上腺素、尿去甲肾上腺素、尿多巴胺、血浆间甲肾上腺素、血浆去甲变肾上腺素均正常。精液常规:无精子症。影像学检查:肾上腺MRI:双侧肾上腺增粗,左侧肾上腺区结节(图1),考虑双侧肾上腺增生。垂体MRI检查结果未见异常。阴囊彩色超声检查结果:左侧精索静脉曲张,双侧睾丸体积偏小(左侧睾丸2.6×1.5 cm、右侧睾丸2.5×1.4 cm)。肾素-醛固酮检查结果:肾素、血醛固酮、醛固酮/肾素比值均正常。快速ACTH兴奋试验结果:ACTH注射后血皮质醇水平无明显升高,提示肾上腺皮质功能减退,糖皮质激素储备功能不足。人绒毛膜促性腺激素(HCG)刺激试验(三日法)结果提示雄激素合成酶缺陷。胰岛素低血糖-促性腺激素释放激素(GnRH)复合刺激试验见表1。低血糖刺激后生长激素(GH)>10 μg/L,可排除生长激素缺乏症,但由于长期高水平雄烯二酮和硫酸去氢表雄酮,反馈性抑制促黄体激素(LH)、促卵泡激素(FSH),导致垂体惰性。染色体核型分析:核型46XY。21-羟化酶(CYP21)基因检测结果提示患者CYP21A2基因第2号内含子存在I2g纯合突变(图2)。结合患者病史和检查结果,考虑诊断为CYP21缺乏症(21-OHD)。治疗给予强的松5 mg每日1次口服。患者出院后未复诊,后失访。

表1 胰岛素低血糖-GnRH复合刺激试验结果
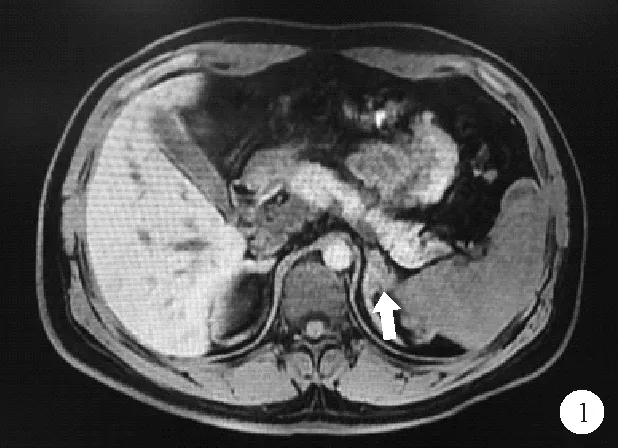
图1 患者2018年10月27日肾上腺MRI结果:双侧肾上腺增粗,左侧肾上腺区结节,如箭头所示
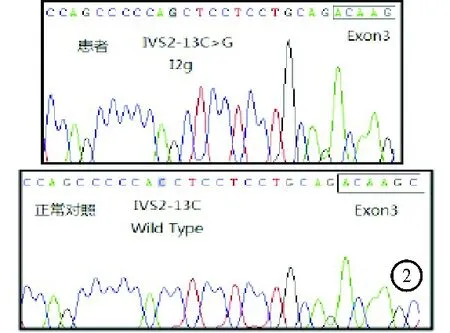
图2 患者CYP21基因检测结果
讨 论
21-OHD是先天性肾上腺增生症中最常见的一类,约占先天性肾上腺增生症的95%[1],主要是由于CYP21的缺乏导致肾上腺糖皮质激素和(或)肾上腺盐皮质激素合成障碍而反馈性引起ACTH分泌增加,从而导致肾上腺增生、肾上腺来源的雄激素增多[2]。在皮质激素合成过程中,CYP21分别催化孕酮和17-羟孕酮使其分别转化为11-脱氧皮质酮和11-脱氧皮质醇,后者再经11-β羟化酶转化分皮质酮和皮质醇[3]。由于CYP21功能缺乏或不足,导致糖皮质激素和盐皮质激素不足,而雄激素合成增加。21-OHD根据临床表现和激素水平可分为经典型(失盐型和单纯男性化型)和非经典型。21-OHD是常染色体隐性遗传疾病,但女性发病率明显高于男性[2]。
21-OHD与CYP21等位基因有关。分子遗传分析结果表明,CYP21A2基因的突变常由基因置换、大规模基因片段缺失和不平等的交换所致[4]。常见的突变包括P30L、V281L突变、2号外显子点突变等[5]。P30L、V281L突变常见于非经典型21-OHD,2号外显子点突变常见于经典型21-OHD[5]。New等[6]对440例非经典型21-OHD的患者研究发现,21-OHD的突变基因型与其表型有一定的相关性,约99%的非经典型21-OHD患者基因型与表型一致,仅1%的非经典型21-OHD患者基因型和表型不一致。本例患者即是临床表型和基因型不一致,其临床表现为非经典型,然而基因检测为2号内含子点,由C突变为G。
21-OHD的临床表现和严重程度与CYP21等位基因变异导致CYP21活性降低的程度有关[3,5]。失盐型21-OHD其酶活性完全消失,新生儿在出生2周内即可出现肾上腺危象危及生命[3]。单纯男性化型21-OHD其酶活性仍保留1%~2%,可合成少量的醛固酮而避免新生儿肾上腺危象的出现[5,7]。非经典型21-OHD其酶活性可保留20%~60%[5],故临床症状较轻。本例患者8岁左右出现第二性征发育,身高明显高于同龄儿童,成年后身高明显低于与同龄人,提示存在性早熟。患者因男性第二性征和性功能没有明显异常,一直到婚后因“不育症”才就诊,发现“无精症”。起病比较隐匿,成年后才得以确诊。本病例还需与松果体钙化相鉴别,松果体可分泌褪黑素,松果体钙化导致褪黑素分泌异常时患者常出现睡眠障碍,并伴有头痛等神经系统疾病[8];褪黑素分泌异常还可导致下丘脑-垂体-性腺轴分泌异常而表现出性早熟,性早熟常见于儿童或年轻人[8-9]。非经典21-OHD患者主要表现为儿童时期的性早熟,骨龄超前,骨骺过早闭合而影响最终身高,男性患者通常其性功能不受影响,但雄激素过高可导致不育,因是男性患者,起病更为隐匿,成年后才得以就诊。女性患者则可出现性功能减退和月经紊乱如闭经、月经稀发、不孕等,相比而言,在青春期有临床表现相对易确诊[4,6]。
男性非经典21-OHD患者肾上腺来源的雄激素过多而引起的症状较轻,往往不易被发现,常因成年后不育而就诊。目前有研究认为造成男性21-OHD患者生育能力下降的最主要因素是睾丸肾上腺残余瘤(TARTs),其次是促性腺激素分泌被负反馈抑制,这两种因素均可导致睾丸功能的损伤,最终影响生精功能[10]。而睾丸肾上腺残余瘤是男性21-OHD患者不育和性腺功能受损的常见原因[11]。通过Pubmed查阅男性21-OHD不育病例报道,绝大多数患者都存在睾丸肾上腺残余瘤。而这些报道中单纯的21-OHD无睾丸残余瘤患者仅有数例报道。本例为非经典21-OHD患者且无睾丸残余瘤。
Bouvattier等[12]对2011~2014年间确诊的219例男性21-OHD患者分析发现无精症患者约占42%。而另一项研究发现,在先天性肾上腺增生的男性患者中,与正常成年男子相比,精子的整体参数较差,而男性非经典21-OHD患者治疗不育的目标主要是改善精子质量[13]。对于非经典21-OHD患者应尽早补充肾上腺皮质激素,以抑制ACTH的分泌,从而抑制肾上腺分泌过多的雄激素,使促性激素分泌正常,恢复睾丸生精和卵巢排卵功能。青少年和成人非经典21-OHD应用糖皮质激素替代治疗,如强的松(每日5.0~7.5 mg,分1~2次口服)、强的松龙(每日3~7 mg,分1~2次口服)或地塞米松(每日0.25~0.50 mg口服)[14]。糖皮质激素治疗过程中需注意患者性激素水平,随时调节糖皮质激素的剂量。
目前国内外关于非经典21-OHD的病例报道并不少见,但主要以儿童、成年女性多见,成年男性的报道相对较少。本例患者初诊时以“无精症”就诊于泌尿外科,后转内分泌科经过一系列的检查排除,确诊为非经典21-OHD。非经典21-OHD的男性患者由于出生时没有典型的外生殖器异常表现而易漏诊,大多数患者因成年后身高低于同龄人或不育而就诊。通过对本病例的报道及相关文献的复习,建议在成年男性不育患者的诊疗过程中提高对21-OHD的认识,可避免误诊及漏诊,同时能予以早期干预提高患者生活质量。
