茶芎HPLC指纹图谱的建立及差异性成分的含量测定Δ
2022-08-26龚伟伟罗光明秦倩曾金祥徐葱茏刘明贵张寿文江西中医药大学药学院南昌330004江西景德中药股份有限公司江西九江33000
龚伟伟,罗光明,秦倩,曾金祥,徐葱茏,刘明贵,张寿文#(.江西中医药大学药学院,南昌 330004;.江西景德中药股份有限公司,江西九江 33000)
伞形科植物茶芎Ligusticum sinenseOliv.cv.Chaxiong为一年生草本植物,以根茎入药,主产于我国江西武宁、瑞昌、德安等地。据《江西省中药材标准》记载,茶芎味辛、性温,归肝、心经,有活血行气、祛风止痛的功效,可用于月经不调、痛经、闭经、产后瘀阻腹痛、胸胁刺痛、头痛、风湿痹痛等[1]。现代研究表明,茶芎主要含有苯酞类、有机酸类、多糖类、氨基酸类和挥发油等成分。其中,苯酞类物质包括洋川芎内酯Ⅰ、洋川芎内酯A和藁本内酯等,均有较好的抗炎、镇痛作用,尤以藁本内酯的作用最为显著;有机酸类物质包括绿原酸、阿魏酸等,绿原酸可调节机体物质代谢、降低胆固醇,还具有抗氧化、抗菌作用,阿魏酸有抗血栓形成、促进血管舒张、延缓动脉粥样硬化的作用[2-6]。可见,上述成分可能是茶芎的活性成分。
目前,关于中药质量评价和质量控制研究多集中于单一或少数成分的定量分析,难以全面、客观地对药材进行评价,因此需要建立全面、有效的质量评价方法。中药指纹图谱能快速反映其化学成分信息,定性判断其真实性;多元统计分析[聚类分析(cluster analysis,CA)、主成分分析(principal component analysis,PCA)、正交偏最小二乘-判别分析(orthogonal partial least squares-discriminant analysis,OPLS-DA)]可对已有的化学成分信息进行定量评价[7-10]。可见,中药指纹图谱与多元统计分析相结合,可全面、有效地对药材进行质量评价和质量控制。基于此,本研究拟采用高效液相色谱(HPLC)法建立12批茶芎药材的指纹图谱并进行相似度分析;同时,拟采用CA、PCA、OPLS-DA法对所得图谱信息进行分析,筛选影响药材质量的差异性成分并对其进行含量测定,旨在为全面进行茶芎质量评价和质量控制提供参考。
1 材料
1.1 主要仪器
本文所用主要仪器包括LC-20AT型HPLC仪(日本Shimadzu公司)、LT-DHC160型高速离心机[立德泰勀(上海)科学仪器有限公司]、KQ-200KDB型高功率数控超声波清洗器(昆山市超声仪器有限公司)、MS205DU型电子分析天平[梅特勒托利多科技(中国)有限公司]等。
1.2 主要药品与试剂
E-藁本内酯对照品(批号Z812557,纯度≥98%)购自上海楷森生物科技有限公司;绿原酸对照品(批号327-97-9,纯度≥98%)、阿魏酸松柏酯对照品(批号63644-62-2,纯度≥98%)、洋川芎内酯A对照品(批号Y08301903021,纯度≥98%)、洋川芎内酯Ⅰ对照品(批号Y08501904030,纯度≥98%)、Z-藁本内酯对照品(批号PCS-201104,纯度≥98%)均购自北京中科质检生物技术有限公司;阿魏酸对照品(批号110773-201915,纯度≥99.4%)购自中国食品药品检定研究院;乙腈为色谱纯,甲醇、磷酸为分析纯,水为超纯水。
12批茶芎药材分别采自江西武宁、瑞昌、德安的3个中药基地,经江西中医药大学药学院付小梅教授鉴定均为茶芎L.sinenseOliv.cv.Chaxiong的成熟根茎。将所采药材放至55℃烘箱中烘干,粉碎,过四号筛,密封保存,备用。12批茶芎药材样品的来源信息见表1。
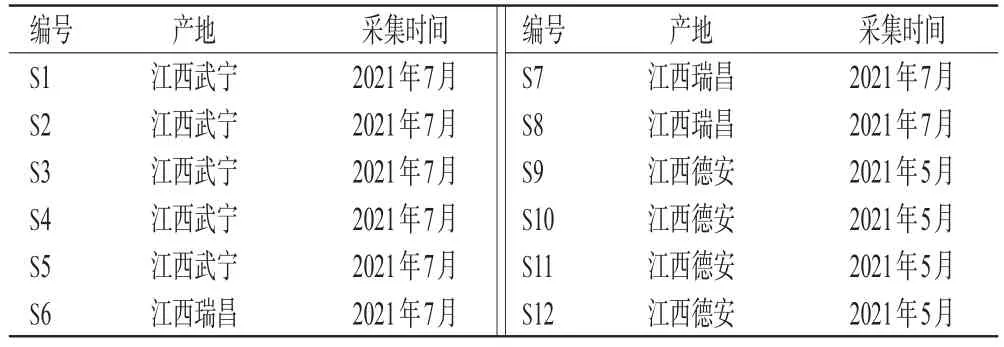
表1 12批茶芎药材样品的来源信息
2 方法与结果
2.1 茶芎药材HPLC指纹图谱的建立
2.1.1 色谱条件以Venusil MP C18(250 mm×4.6 mm,5 μm)为色谱柱,以0.1%磷酸溶液(A)-乙腈(B)为流动相进行梯度洗脱(0~10 min,10%B→15%B;10~13 min,15%B;13~23 min,15%B→20%B;23~26 min,20%B;26~36 min,20%B→30%B;36~39 min,30%B;39~47 min,30%B→40%B;47~53 min,40%B→55%B;53~60 min,55%B→65%B;60~75 min,65%B);检测波长为280 nm;流速为1.0 mL/min;柱温为30℃;进样量为10 μL;检测平衡时间为20 min。
2.1.2 混合对照品溶液的制备 取绿原酸、阿魏酸、洋川芎内酯A、洋川芎内酯Ⅰ、阿魏酸松柏酯、E-藁本内酯、Z-藁本内酯对照品各适量,分别置于10 mL棕色量瓶中,加70%甲醇溶解并定容,即得各单一对照品贮备液。分别取上述各单一对照品贮备液适量,置于同一10 mL棕色量瓶中,加70%甲醇定容,制成上述成分质量浓度分别为56.3、42.5、40.5、53.9、33.7、23.1、96.1 μg/mL的混合对照品溶液。
2.1.3 供试品溶液的制备 取茶芎样品粉末约0.5 g,精密称定,置具塞锥形瓶中,加70%甲醇25 mL,称定质量,超声(功率200 W,频率40 kHz)提取30 min,放冷,再次称定质量,用70%甲醇补足减失的质量,摇匀,以13 000 r/min离心5 min,取上清液,用0.45 μm微孔滤膜滤过,取续滤液,即得。
2.1.4 精密度试验 取茶芎样品(编号S1)粉末,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件连续进样测定6次,以Z-藁本内酯(峰形、分离度均较好且响应较强,下同)为参照,计算各共有峰的相对保留时间和相对峰面积。结果显示,各共有峰相对保留时间和相对峰面积的RSD均小于3.0%(n=6),表明方法精密度良好。
2.1.5 重复性试验 取茶芎样品(编号S1)粉末6份,分别按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定,以Z-藁本内酯为参照,计算各共有峰的相对保留时间和相对峰面积。结果显示,各共有峰相对保留时间和相对峰面积的RSD均小于3.0%(n=6),表明方法重复性良好。
2.1.6 稳定性试验 取茶芎样品(编号S1)粉末,按“2.1.3”项下方法制备供试品溶液,分别于室温下放置0、2、4、8、12、24 h时按“2.1.1”项下色谱条件进样测定,以Z-藁本内酯为参照,计算各共有峰的相对保留时间和相对峰面积。结果显示,各共有峰相对保留时间和相对峰面积的RSD均小于3.0%(n=6),说明该供试品溶液在室温下放置24 h内稳定性良好。
2.1.7 指纹图谱的建立 取12批茶芎样品,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定,记录HPLC图并以“AIA”格式导入《中药色谱指纹图谱相似度评价系统(2012版)》。以S7样品图谱(所含色谱信息丰富且各共有峰的分离度均较好)为参照,经多点校正、Mark峰匹配后,生成茶芎HPLC叠加指纹图谱和对照指纹图谱(R),详见图1。
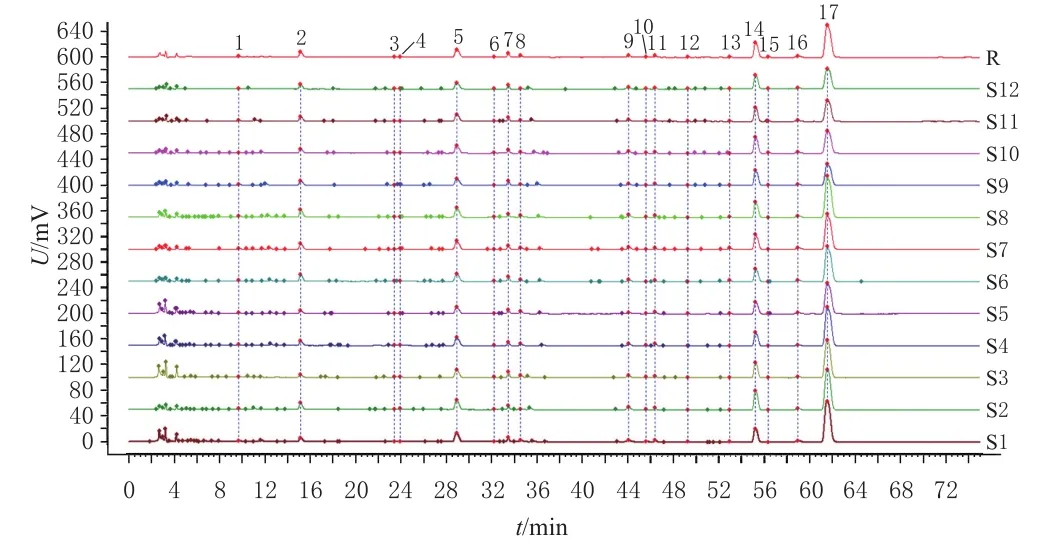
图1 12批茶芎样品的HPLC叠加指纹图谱和对照指纹图谱
2.1.8 共有峰的确定及指认 采用Mark峰匹配,共得到17个共有峰。通过查阅相关文献[11-12]并比对混合对照品色谱图(图2),初步确定1号峰为绿原酸、2号峰为阿魏酸、7号峰为洋川芎内酯Ⅰ、9号峰为阿魏酸松柏酯、13号峰为E-藁本内酯、14号峰为洋川芎内酯A、17号峰为Z-藁本内酯。

图2 混合对照品溶液的HPLC图
2.1.9 相似度评价 使用《中药色谱指纹图谱相似度评价系统(2012版)》,以对照指纹图谱为参照,对12批茶芎样品进行相似度评价。结果显示,S1~S12批样品与对照指纹图谱的相似度分别为0.997、1.000、0.999、0.997、0.998、0.998、1.000、0.998、0.989、0.989、0.991、0.990,均大于0.980,提示不同产地茶芎样品的相似性较好,质量相对稳定。其中,武宁茶芎(S1~S5)的相似度为0.997~1.000,瑞昌茶芎(S6~S8)的相似度为0.998~1.000,德安茶芎(S9~S12)的相似度为0.989~0.991。整体上,武宁茶芎(S1~S5)与瑞昌茶芎(S6~S8)的相似度接近,提示两者化学成分相似;此外,武宁、瑞昌茶芎的相似度均稍高于德安茶芎(S9~S12),提示德安茶芎的化学成分可能与前两者有所不同。
2.2 茶芎样品的CA
本研究以12批茶芎样品17个共有峰的绝对峰面积为变量,使用SPSS 22.0软件进行Z-score标准化处理后,采用平方欧氏距离为测度,以组间连接法进行CA,结果见图3。由图3可知,当平方欧氏距离为20时,12批茶芎样品可被分为3类,S1~S5(武宁茶芎)聚为一类,S6~S8(瑞昌茶芎)聚为一类,S9~S12(德安茶芎)聚为一类。由此可以看出,不同产地茶芎样品具有较明显的聚类趋势。当平方欧氏距离为24时,S1~S5(武宁茶芎)可与S6~S8(瑞昌茶芎)聚为一类,S9~S12(德安茶芎)聚为一类,与茶芎HPLC指纹图谱相似度评价结果基本一致。
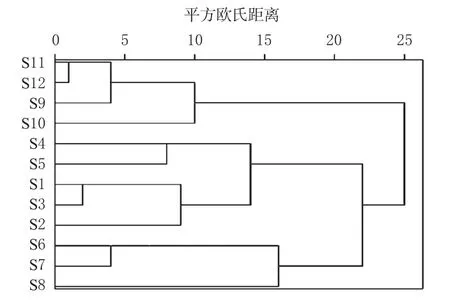
图3 12批茶芎样品的CA树状图
2.3 茶芎样品的PCA
本研究以12批茶芎样品17个共有峰的绝对峰面积为变量,采用SPSS 22.0软件,在进行KMO、Bartlett’s检验和降维统计的基础上进行PCA,其总方差解释结果见表2。由表2可知,本研究共提取了4个主成分,累计方差贡献率为85.385%(>85%),提示前4个主成分代表了85.385%的整体信息,可用于反映不同产地茶芎样品的整体质量。
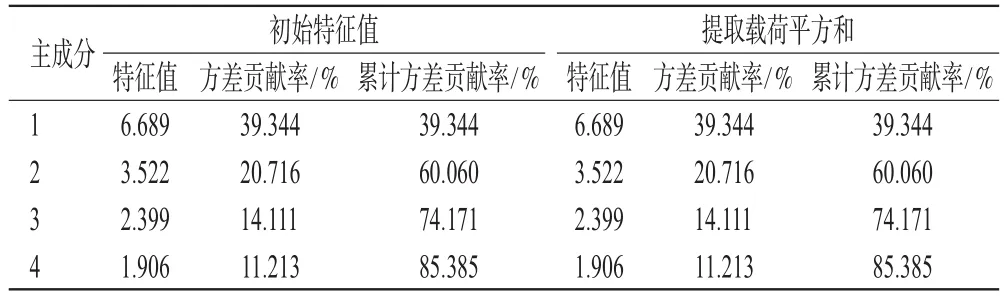
表2 总方差解释结果
由因子矩阵和权重系数分析可知,主成分1的信息主要来自2、8、10、12、13、16号峰,主成分2的信息主要来自11、14、15、17号峰,主成分3的信息主要来自3、4、6、7号峰,主成分4的信息主要来自1、5、9号峰。对各因子进行标准化处理,结合上述因子矩阵及权重系数,得到反映茶芎样品质量的主成分得分(F1~F4)表达式:

随后,对F1~F4进行系数加权(加权系数为特征值与累计特征值之和的比值),得到综合评分(Fz)的表达式:
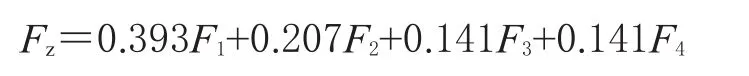
综合评分越高,表示该批样品整体质量越优[13-14]。根据上述公式,对12批不同产地茶芎样品的质量进行评价,结果见表3。由表3可知,瑞昌茶芎(S6~S8)质量较好,其次为武宁茶芎(S1~S5)和德安茶芎(S9~S12)。
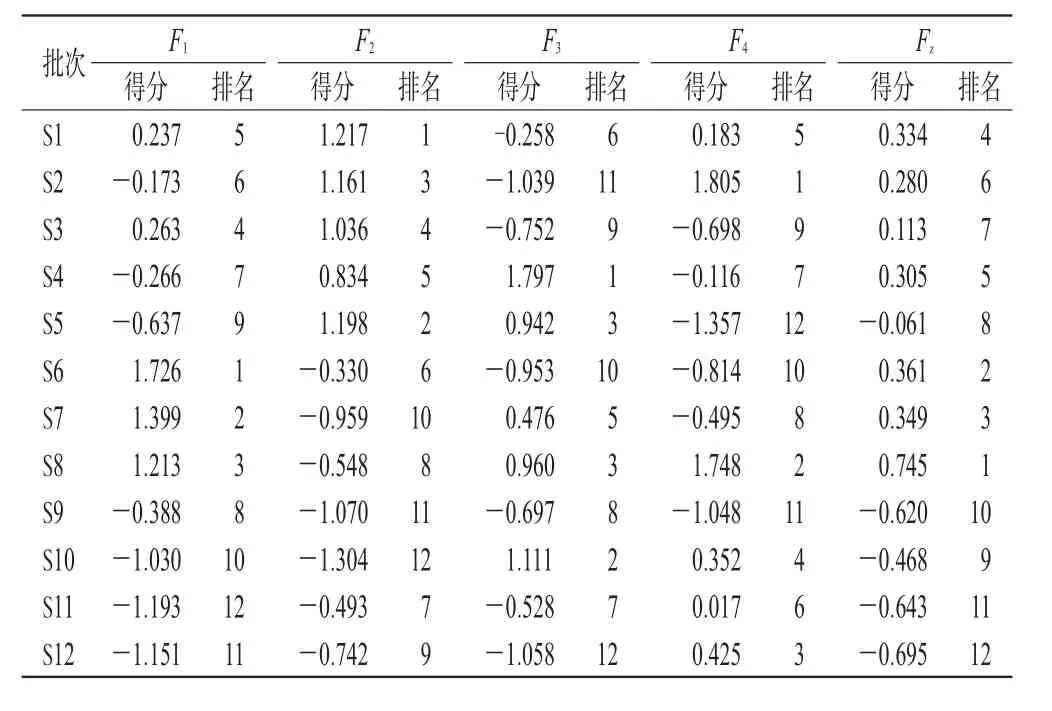
表3 主成分综合评分及排序
2.4 茶芎样品的OPLS-DA
本研究以12批茶芎样品17个共有峰的绝对峰面积为变量,使用SIMCA 14.1软件进行OPLS-DA。结果显示,X轴方向的模型解释率(R2X)为0.945,Y轴方向的模型解释率(R2Y)为0.987(R2X、R2Y越接近1,表明模型的稳定性越强[15-16]),模型的预测率(Q2)为0.966(Q2越接近1,表明模型的可预测性越强[15-16])。随机排列200次进行置换检验验证,判断OPLS-DA模型有无过度拟合现象。结果显示,所得模型的R2截距为0.274,Q2截距为-1.290,提示所建模型不存在过度拟合现象[15-16],可用以有效区分不同产地的茶芎样品。由OPLS-DA得分图(图4)可知,武宁茶芎(S1~S5)聚为一类,瑞昌茶芎(S6~S8)聚为一类,均分布在得分图右侧;德安茶芎(S9~S12)聚为一类,分布在得分图左侧,该结果与相似度评价、CA结果基本一致,可相互佐证。

图4 OPLS-DA得分图
进一步对OPLS-DA模型的变量重要性投影(variable importance in the projection,VIP)进行分析,筛选影响样品质量的差异性成分。以VIP>1为筛选标准[16],共得到贡献相对较大的4个差异性成分,依次为17号峰(Z-藁本内酯,VIP为3.174)、14号峰(洋川芎内酯A,VIP为1.195)、2号峰(阿魏酸,VIP为1.150)、13号峰(E-藁本内酯,VIP为1.052)。结果见图5。
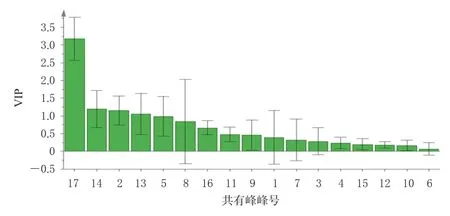
图5 共有峰的VIP图
2.5 4种差异性成分的含量测定
采用HPLC法对“2.4”项下所得4种差异性成分(阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯)的含量进行测定。
2.5.1 色谱条件 同“2.1.1”项。
2.5.2 混合对照品溶液的制备 取阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯4种对照品各适量,按“2.1.2”项下方法制备各单一对照品贮备液。取上述各单一对照品贮备液适量,按“2.1.2”项下方法制成上述成分质量浓度分别为1.84、1.96、1.68、2.98 mg/mL的混合对照品溶液。
2.5.3 供试品溶液的制备 同“2.1.3”项。
2.5.4 专属性试验 取“2.5.2”项下混合对照品溶液、“2.5.3”项下供试品溶液和空白对照溶液(70%甲醇),按“2.5.1”项下色谱条件下进样测定,记录色谱图(图略)。结果显示,阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯色谱峰的分离度均大于1.5,且各色谱峰无拖尾现象,提示方法专属性良好。
2.5.5 线性范围考察 取“2.5.2”项下阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯混合对照品溶液0.1、0.5、1、2、5 mL,分别置于10 mL量瓶中,加70%甲醇定容,再按“2.5.1”项下色谱条件进样测定,记录峰面积。以待测成分的质量浓度为横坐标(X)、峰面积为纵坐标(Y)进行线性回归,结果见表4。分别取阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯混合对照品溶液(质量浓度分别为0.018、0.020、0.017、0.030 mg/mL,按“2.5.2”项下方法配制)适量,以70%甲醇倍比稀释,按“2.5.1”项下色谱条件进样测定,分别以信噪比3∶1、10∶1计算检测限和定量限,结果见表4。
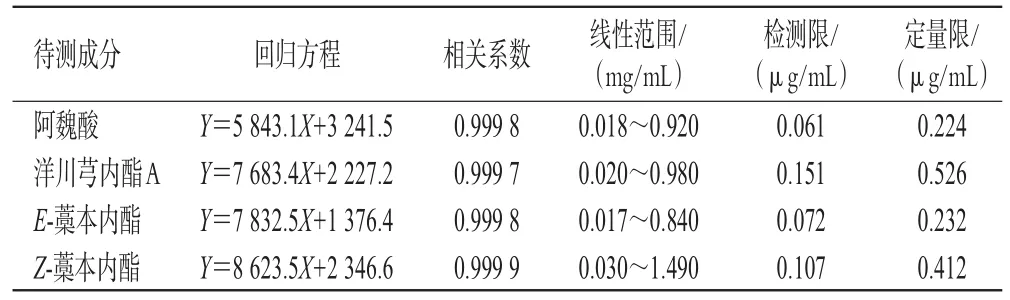
表4 4种成分的线性关系考察结果
2.5.6 精密度试验 取“2.5.3”项下供试品溶液(编号S1),按“2.5.1”项下色谱条件连续进样6次,记录峰面积。结果显示,阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯峰面积的RSD分别为0.12%、0.54%、0.98%、0.42%(n=6),表明方法精密度良好。
2.5.7 重复性试验 取同一批茶芎样品(编号S1)6份,按“2.5.3”项下方法制备供试品溶液,再按“2.5.1”项下色谱条件进样测定,记录峰面积并按外标法计算含量。结果显示,阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯含量的RSD分别为0.93%、1.38%、1.45%、1.12%(n=6),表明方法重复性良好。
2.5.8 稳定性试验 取“2.5.3”项下同一供试品溶液(编号S1),分别于室温下放置0、2、4、8、12、24 h时按“2.5.1”项下色谱条件进样测定,记录峰面积。结果显示,阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯峰面积的RSD分别为0.18%、0.17%、0.50%、0.25(n=6),表明该供试品溶液在室温下放置24 h内稳定性良好。
2.5.9 加样回收率试验 取已知含量的样品(编号S4)6份,每份约0.25 g,置具塞锥形瓶中,加入与已知量相当的阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯对照品溶液(按“2.5.2”项下方法配制),按“2.5.3”项下方法制备供试品溶液,再按“2.5.1”项下色谱条件进样测定,记录峰面积并计算加样回收率。结果显示,阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯的平均加样回收率分别为99.92%、98.01%、100.90%、99.73%,RSD分别为0.69%、0.26%、1.45%、0.17%(n=6)。
2.5.10 含量测定 分别取12批茶芎样品,按“2.5.3”项下方法制备供试品溶液,再按“2.5.1”项下色谱条件进样测定,记录峰面积并按外标法计算含量。采用Graph-Pad Prism 9.3.0软件对数据进行统计分析,组间比较采用单因素方差分析,检验水准α=0.05,结果见表5。由表5可知,12批茶芎药材中,阿魏酸含量均在0.4 mg/g以上,3个产地茶芎中该成分含量两两比较差异均有统计学意义,且以瑞昌茶芎最高(P<0.05或P<0.01),故可作为标志性成分;洋川芎内酯A含量均高于或接近于1.2 mg/g,3个产地茶芎中该成分含量两两比较差异均有统计学意义,且以瑞昌茶芎最高(P<0.05或P<0.01),故也可作为标志性成分;E-藁本内酯含量均在0.1 mg/g以上,两两比较差异显著且德安茶芎显著高于其他产地茶芎(P<0.01),故也可作为标志性成分;瑞昌、武宁、德安茶芎中Z-藁本内酯的含量分别高于8.0、7.0、4.0 mg/g,两两比较差异均有统计学意义(P<0.01),故也可作为标志性成分。上述含量测定结果也佐证了OPLS-DA模型的预测结果,阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯均可作为影响质量的差异性成分。
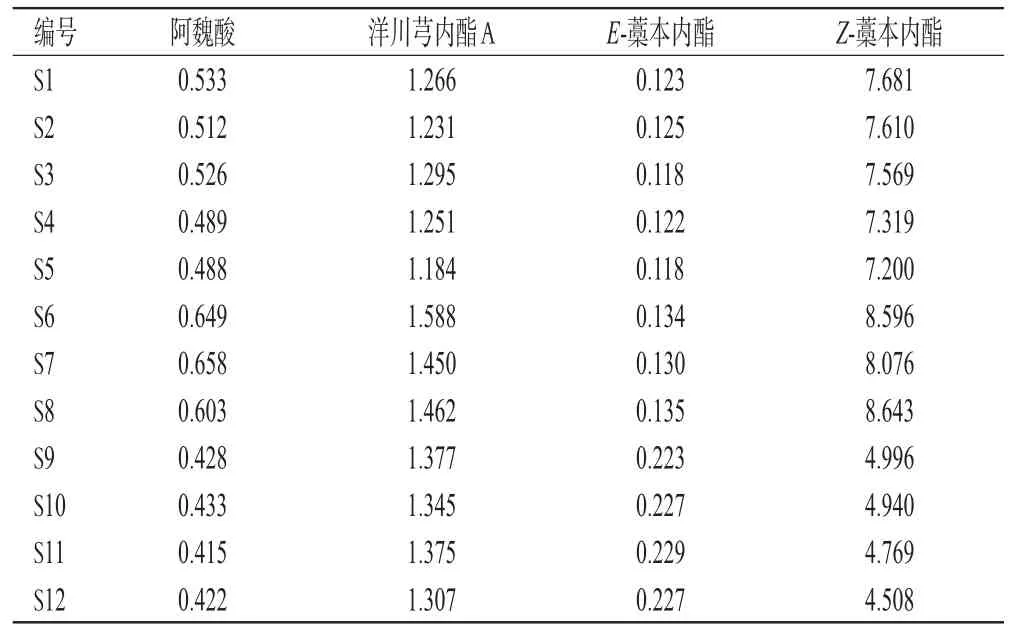
表5 12批茶芎样品中4种成分的含量测定结果(mg/g)
3 讨论
3.1 提取溶剂及提取方式的选择
在前期预实验中,本课题组分别考察了不同提取溶剂(水、30%甲醇、50%甲醇、70%甲醇、甲醇)对目标成分含量的影响,经单因素方差分析可知,70%甲醇对目标成分的提取效果最好,且与其他溶剂提取效果比较,差异均有统计学意义(P<0.05)。同时,本课题组分别考察了不同提取方式(回流提取法、超声提取法)对目标成分含量的影响,经单因素方差分析可知,两种提取方式对目标成分含量的影响并不显著(P>0.05),故考虑到实验操作的便捷性,本研究选择了超声提取法。
3.2 流动相及检测波长的选择
在色谱条件考察过程中,由于样品含有有机酸类成分,为防止色谱峰拖尾,故使用0.1%磷酸溶液作为水相,并先后考察了乙腈-0.1%磷酸溶液和甲醇-0.1%磷酸溶液的洗脱效果。结果显示,当以乙腈-0.1%磷酸溶液为流动相时,所得色谱图基线较为平稳,各色谱峰峰形和分离度均较好,故选择乙腈-0.1%磷酸溶液为流动相。通过二极管阵列紫外-可见光检测器在190~800 nm内进行全波长扫描发现,当吸收波长为280 nm时,各色谱峰峰形和分离度均较好,参照峰(Z-藁本内酯)响应较强,故最终选择检测波长为280 nm。
3.3 指纹图谱及多元统计分析
12批茶芎药材共有17个共有峰。相似度评价结果显示,12批茶芎药材的相似度均高于0.980,说明3个产地茶芎药材的化学成分种类差异较小,药材质量相对稳定;根据CA和OPLS-DA结果可知,S1~S5(武宁茶芎)聚为一类,S6~S8(瑞昌茶芎)聚为一类,S9~S12(德安茶芎)聚为一类,提示药材受产地环境的影响较为明显。根据PCA综合评价分析可知,S6~S8(瑞昌茶芎)的综合评分较高,其次为武宁茶芎(S1~S5)和德安茶芎(S9~S12)。可见,CA、PCA、OPLS-DA可为不同产地茶芎药材的质量评价和质量控制提供依据。
3.4 差异性成分的筛选及含量测定分析
中药成分具有复杂性和多效性,采用单一指标成分难以实现对其质量的整体控制,故多种成分的同时测定是中药质量控制的重要研究方向。本研究运用OPLS-DA筛选出4个差异性成分,即阿魏酸、洋川芎内酯A、E-藁本内酯、Z-藁本内酯。通过含量测定发现,3个产地茶芎样品中4种成分两两比较,差异均有统计学意义,再次说明这4种成分可作为标志性成分用以区分不同产地的茶芎药材,并可为该药材的全面评价和质量控制提供参考。
综上所述,本研究所建指纹图谱、多元统计分析、含量测定方法稳定、可行,可用于茶芎药材评价和质量控制。阿魏酸、洋川芎内酯A、Z-藁本内酯、E-藁本内酯可能是影响不同产地茶芎药材质量的差异性成分,且前3种成分含量均以瑞昌茶芎最高,E-藁本内酯含量以德安茶芎最高。
