RGO/Al复合材料界面性质第一性原理研究
2022-08-25孙铭邵溥真孙凯黄建华张强修子扬肖海英武高辉
孙铭, 邵溥真, 孙凯, 黄建华, 张强, 修子扬, 肖海英, 武高辉
RGO/Al复合材料界面性质第一性原理研究
孙铭, 邵溥真, 孙凯, 黄建华, 张强, 修子扬, 肖海英, 武高辉
(哈尔滨工业大学 材料科学与工程学院, 哈尔滨 150001)
本研究采用基于密度泛函理论的第一性原理方法, 在广义梯度近似下, 分别建立了具有不同碳氧比的“铝/氧化石墨烯/铝(Al/GO/Al)”界面模型以及含缺陷“Al/GO/Al”三层界面模型。探讨了含氧官能团和单空位缺陷、双空位缺陷以及拓扑缺陷对还原氧化石墨烯增强铝基复合材料界面性质的影响。研究结果表明: 在“Al/GO/Al”界面模型中, 环氧基优于碳原子而与铝原子产生明显的电荷交互作用, 氧原子净电荷为–0.98 e, 铝原子净电荷为0.46 e, 环氧基有利于复合材料中还原氧化石墨烯与铝基体之间的界面结合。当缺陷存在时, 含缺陷的“Al/GO/Al”界面模型中缺陷处碳原子净电荷在–0.05 e至–0.38 e区间, 环氧基与碳原子之间存在较弱的相互作用, 与铝原子间相互作用明显较强。环氧基抑制了空位缺陷处碳原子与铝原子之间的反应, 可保护含空位还原氧化石墨烯中碳原子结构的完整性。本研究可为开发高性能Al/GO/Al基复合材料提供理论指导。
第一性原理; 还原氧化石墨烯/铝复合材料; 氧化石墨烯/铝界面模型; 界面性质
铝基复合材料以较高的比强度、良好的尺寸稳定性和易加工成型等优异的性能, 而被广泛应用于航空航天和汽车电子等行业[1-3]。随着航空航天等行业的不断发展, 对高性能铝基复合材料的需求越来越大, 急需对传统铝基复合材料进行改性[4]。石墨烯(Gr)作为一种二维碳纳米材料, 具有强度高、延展性好、导电导热性强等优良性能[5-10], 有望为铝基复合材料的改性提供增强体, 实现所需性能的突破。但由于石墨烯片层间具有范德华力, 且比表面积大, 在制备过程中易产生团聚, 影响复合材料性能[11-15]。为实现石墨烯均匀分散, 常用机械球磨分散和液相分散工艺[16-18]。其中液相分散常用石墨烯的前驱体, 如氧化石墨烯(GO), 在制备过程中分散并还原为还原氧化石墨烯(RGO), 实现复合材料中RGO与基体间的界面设计。然而值得注意的是, 以此方法制得的复合材料中, RGO表面会残留一定程度的含氧官能团[19]。而这一部分残留对复合材料性能, 尤其是界面性能的影响较大, 且难以直接通过实验研究其影响。因此, 有必要通过模拟对RGO/Al复合材料的界面性质进行研究。
基于密度泛函理论(DFT)的第一性原理方法对复合材料界面性质的研究已经取得了一些进展[20-23]。其中, Zhang等[24]研究了缺陷对石墨烯/铝界面结合的影响, 发现缺陷处C–Al之间形成的较强共价键是提升复合材料力学性能的重要原因。但采用第一性原理研究含氧官能团对RGO/Al界面性质的影响则尚未见报道。
本工作运用基于密度泛函理论的第一性原理方法, 模拟研究不同碳氧比的铝/氧化石墨烯/铝(Al/GO/Al)界面模型, 探讨含缺陷Al/GO/Al界面模型的界面性质, 分析计算两类模型的界面原子结构、界面能、界面粘附功以及界面电子结构, 从微观层面揭示RGO/Al复合材料界面处C–Al原子之间的交互作用, 揭示含氧官能团与缺陷对界面性质的影响, 为开发高性能Al/GO/Al基复合材料提供指导。
1 计算方法和模型
1.1 计算方法
采用基于DFT第一性原理的平面波赝势法, 利用Material Studio中的CASTEP程序实现模型结构优化、能量计算以及电子结构分析。选用Ultrasoft超软赝势, 交换关联能量泛函采用广义梯度近似中的Perdew-Burke-Ernzerhof形式, 同时考虑到范德华力, 计算时选用Grimme色散校正(DFT-D correction), 在实空间内进行计算。平面波截断能采用420 eV, 布里渊区k点取样网格设置为6×6×1, 总能收敛标准优于2.0×106eV。
1.2 结构模型
GO是Gr的一种前驱体, 具有典型的准二维空间结构, 其片层上有大量的羟基、羧基以及环氧基等含氧官能团, 因制备工艺不同, 碳表面的含氧官能团也存在一定程度的不同[25-26]。为了简化模型并便于对比, 在模型建立时假设含氧官能团均为环氧基, 位置随机分布。图1(a~e)为结构优化后的Gr原子模型和不同碳氧比的GO原子模型。由于C原子与O原子之间存在相互作用, GO表面存在褶皱。
本工作建立了含有一个环氧基与单空位缺陷(Single-vacancy defect, SV)、双空位缺陷(Double- vacancy defect, DV)以及拓扑缺陷(Stone-Wales defect, SW)的GO原子模型(图2(a~c))。为便于表述, 对模型中缺陷处部分C原子进行编号。按照与上述界面模型一致的参数构建了图2(d~f)中的含缺陷Al/GO/Al界面模型。

图1 GO(Gr)原子模型与Al/GO(Gr)/Al界面模型
(a) Gr atom model; (b-e) GO atomic models with C/O ratios of 24 : 1, 12 : 1, 8 : 1, and 6 : 1, respectively; (f) Al/Gr/Al interface model; (g-j) Al/GO/Al interface models with C/O ratios of 24 : 1, 12 : 1, 8:1, and 6 : 1, respectively

图2 含缺陷GO原子模型与含缺陷Al/GO/Al界面模型
(a) GO with single-vacancy defect; (b) GO with double-vacancy defect; (c) GO with Stone-Wales defect; (d) Al/GO/Al interface model with single-vacancy defect; (e) Al/GO/Al interface model with double- vacancy defect; (f) Al/GO/Al interface model with Stone-Wales defect
2 结果与讨论
2.1 结构模型的确定


图3 不同原子层数对应Al表面模型的表面能
2.2 环氧基对RGO/Al界面性质的影响
2.2.1 界面能量计算
复合材料的界面结合效果通常采用界面能与界面粘附功进行衡量[23,27]。其中, 界面的稳定性用界面能int评价, 界面能越小, 界面越稳定。界面能int可表示为:

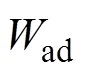

式中,Xslab表示构成界面模型的表面模型X总能量。
Al/GO(Gr)/Al界面模型的界面粘附功与界面能计算结果如表1和图4所示。随界面模型中环氧基数量增加, 界面粘附功增大, 界面能降低, 且与环氧基数量呈线性关系。相比于Gr, GO与Al基体之间的界面结合效果更好, 界面更稳定; 随环氧基数量增多, 界面结合性和稳定性增强, 可见RGO上残留的含氧官能团对界面结合是有利的。文献[28-30]报道, 加入RGO可显著提高RGO/Al复合材料的强度, 可见除了RGO本身具有较高的力学性能外, RGO上残留的含氧官能团可增强界面结合效果, 对提高复合材料力学性能作出一定程度的贡献。
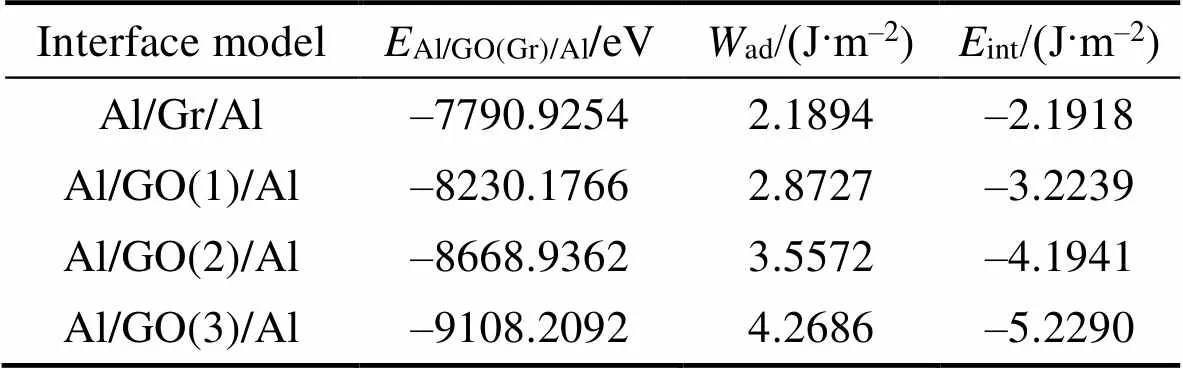
表1 Al/GO(Gr)/Al界面能量计算结果

图4 Al/GO(Gr)/Al界面模型的计算能量随环氧基数量的变化
2.2.2 电子态密度
Al/GO(Gr)/Al界面模型界面层位置的电子分波态密度(PDOS)如图5所示。在Al/Gr/Al与Al/GO/Al界面模型中, C原子的PDOS均无明显变化, 峰位不存在偏移。0 K条件下C原子与Al和O原子的相互作用均不明显; Al原子与O原子在–21 eV附近存在较为明显的共振峰, Al原子的s轨道和p轨道电子均与O原子s轨道发生杂化, 二者之间存在相互作用。随着O原子增多, 共振峰峰强增大, 在–6 eV附近O原子p轨道与Al原子p轨道之间逐渐产生了共振峰。O原子与Al原子之间存在相互作用, 且O原子越多, 相互作用越强。Al与O原子在费米能级以下的峰位随着O原子的增多, 峰位存在向低能量方向移动的趋势, 界面逐渐稳定, 符合界面能量计算的结果。
2.2.3 Mulliken布居分析与差分电荷密度分析
对界面模型进行电子态密度分析可以定性表征界面处原子的电子结构, 通过Mulliken布居分析并与差分电荷密度分析相结合, 可以进一步分析界面处原子成键情况以及电荷转移量[21-22]。含单个环氧基的Al/GO/Al以及Al/Gr/Al模型中界面处原子轨道布居数与差分电荷密度的计算结果分别见表2、表3和图6。表2显示, Al/Gr/Al模型中界面处不同位置Al原子与C原子的布居数基本保持一致, 二者之间电荷转移幅度十分微弱, 净电荷仅为0.02与–0.03 e, 基本不存在相互作用, 这一结果与界面处电子态密度结果相一致。

表2 Al/Gr/Al界面模型Mulliken布居分析

表3 Al/GO/Al界面模型Mulliken布居分析

图6 Al/Gr/Al (a)与Al/GO/Al (b)差分电荷密度
对于Al/GO/Al界面模型而言, Al原子(图6(b)中位置1处铝原子)与界面处距离最近的O原子产生相互作用,Al原子s与p轨道均失去电子, 净电荷为0.46 e, O原子获得电子, 净电荷为–0.98 e, 符合离子键的成键规律, 提升了界面结合强度。距离O原子位置较远的Al原子(图6(b)中位置2处)无显著的电荷转移。此外, 不同位置C原子与O原子的相互作用均不明显, 与分波态密度结果相一致。
综上可知, 在Al/Gr/Al模型中Al原子与C原子之间无明显相互作用。在Al/GO/Al界面模型中, C原子与Al原子、O原子均无相互作用产生。Al原子与O原子之间存在较强的相互作用, Al原子附近电荷密度降低, O原子附近电荷密度增加, 二者之间存在离子键成分。O原子与Al原子的相互作用使界面粘附功增大, 界面能降低, 有利于GO与Al基体间的界面结合, 从而对RGO/Al复合材料的力学性能产生积极影响。
2.3 环氧基与缺陷对RGO/Al界面性质的影响
2.3.1 界面能量计算
三类含缺陷Al/GO/Al界面模型的界面粘附功与界面能计算结果如图7所示。与文献[24]结果相类似, 缺陷处碳原子与基体产生相互作用, 进而影响界面结合效果。含单空位缺陷Al/GO/Al界面模型的界面粘附功稍低于完整结构Al/GO/Al界面模型, 而含双空位缺陷与拓扑缺陷模型的界面粘附功较完整结构更高。可见双空位缺陷与拓扑缺陷有利于GO与Al基体之间的界面结合, 而单空位缺陷对界面结合存在负面影响。三类缺陷界面模型的界面能均高于完整结构Al/GO/Al界面模型, 说明三类缺陷均降低了界面的稳定性, 拓扑缺陷的影响程度最小。

图7 含缺陷Al/GO/Al界面模型能量计算
SW: Stone-Wales; SV: Single-vacancy; DV: Double-vacancy
2.3.2 电子态密度
图8显示了含有三类缺陷的Al/GO/Al界面模型的分波态密度, 并与含单个环氧基的Al/GO/Al界面模型进行对比。与完整Al/GO/Al模型相比, Al原子s轨道与p轨道电子密度均降低, Al除了与O存在相互作用外, 与C原子产生了相互作用。含不同缺陷的Al/GO/Al模型中O原子PDOS与完整Al/GO/Al模型基本一致。与完整Al/GO/Al模型相比, 含缺陷界面模型中C原子s轨道与p轨道最大电子密度明显降低, p轨道峰型存在变化。另外, 在–11至–1.5 eV区间内含缺陷GO的C原子p轨道峰变宽, C原子之间的相互作用、C原子与Al之间作用规律与完整GO的C原子存在区别。引入缺陷破坏了GO上C原子的结构完整性, 致使缺陷处C原子与界面处Al原子产生相互作用, 该结论与文献中的模拟结果相吻合[8,31]。
2.3.3 Mulliken布居分析与差分电荷密度分析
图9显示了含缺陷Al/GO/Al界面模型中不同坐标位置的差分电荷密度分析结果, 与表4的布居分析结果相结合, 可以进一步定量分析模型界面处的电子结构, 其中图9中原子编号与表4中原子位置相对应。三类模型中Al原子(对应编号Al1)与O原子之间均产生了最强烈的相互作用, Al原子失去电子, 附近电荷密度降低, O原子附近电荷密度增加, 二者之间存在离子键作用, 且得失电子的量也与Al/GO/Al模型相一致, 可以认为不同种类缺陷都不影响Al原子与环氧基之间的相互作用。但由于GO表面存在缺陷, C与Al之间的作用规律与完整Al/GO/Al存在较大区别。三类模型当中缺陷处C原子(对应编号C原子)与附近的Al原子(对应编号Al2)均存在不同程度的相互作用, 单空位处C原子与Al原子相互作用最强, 拓扑缺陷次之, 双空位缺陷处C原子与Al原子相互作用最弱。结合界面原子结构发现, 对于含空位缺陷的Al/GO/Al界面模型, 与C原子相互作用最强的Al原子位于环氧基异侧, 而拓扑缺陷Al/GO/Al界面模型中与C原子相互作用最强的Al原子位于环氧基同侧。此外, 受到缺陷影响, 非缺陷部位C原子与Al之间也存在较微弱的相互作用, C原子与Al原子的得失电子数量存在一定的波动。

图8 不同缺陷Al/GO/Al界面模型中不同原子PDOS曲线
(a) PDOS for Al atom; (b) PDOS for C atom; (c) PDOS for O atom

图9 含缺陷的Al/GO/Al差分电荷密度
(a, b) Single-vacancy defect indirection; (c, d) Single-vacancy defect in vertical Z direction; (e, f) Double-vacancy defect indirection; (g, h) Double-vacancy defect in verticaldirection; (i, j) Stone-Wales defect indirection; (k, l) Stone-sWales defect in verticaldirection
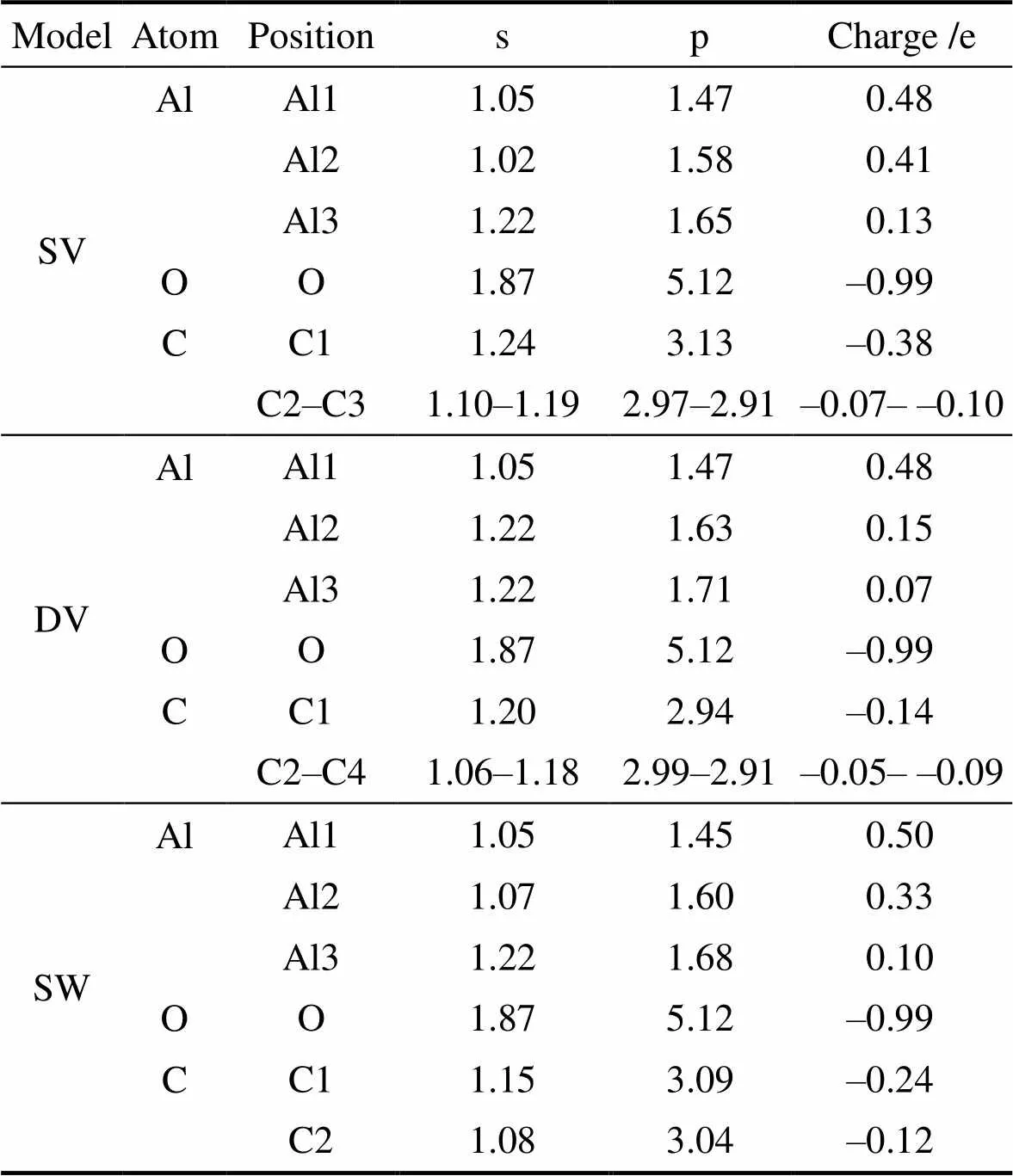
表4 含缺陷Al/GO/Al布居分析
结合界面原子结构、原子轨道布居数以及差分电荷密度结果, 进一步分析含缺陷GO/Al界面模型中C原子的具体作用规律, 结果如图10所示。其中含空位缺陷的两类模型具有相似的规律。由于环氧基, 与C原子相互作用最强的Al原子位于不含环氧基的异侧界面上, 同时空位处与Al原子相互作用最强的C原子始终位于远离环氧基的一侧(图10(a, b)), 单空位缺陷中C原子得到0.38 e, 双空位缺陷中C原子得到0.14 e。而靠近环氧基一侧的C原子与Al原子相互作用相对较弱, 单空位与双空位缺陷中C原子净电荷区间仅为–0.05~–0.10 e。两模型中非空位处C原子同样满足这一规律, 与环氧基相连位置C原子得电子量更少。界面模型中存在的环氧基会抑制C与Al之间的反应, 保护GO中碳原子的结构完整性, 改善界面结合效果, 进而提高RGO/Al复合材料的力学性能。

图10 含缺陷的GO原子模型和相应的差分电荷密度
(a, d) Single-vacancy; (b, e) Double-vacancy; (c, f) Stone-Wales defects
含拓扑缺陷Al/GO/Al界面模型的作用规律与空位缺陷界面模型存在差异。原子结构方面, 与C原子相互作用最强的Al原子位于含有环氧基的同一界面上, 这使得模型中C原子、O原子和Al原子之间均存在相互作用。布居分析与差分电荷密度结果也印证了这一规律。不同于含空位缺陷的界面模型, 拓扑缺陷中与Al原子相互作用最强的C原子位于靠近环氧基一侧, 净电荷为–0.24 e。Al原子中的电子向C、O原子处转移, 最大失电子幅度为0.50 e, 均高于Al原子单独与O原子相互作用的0.46 e以及单独与C原子相互作用的0.48 e。结合能量计算结果, 拓扑缺陷使模型的界面结合效果得到提升。
3 结论
采用基于密度泛函理论的第一性原理计算方法研究了Al/GO(Gr)/Al和含缺陷Al/GO/Al界面模型, 分析了两类模型的界面能量、界面处分波态密度、布居分析以及差分电荷密度等特性, 得到以下结论:
1)GO中距离环氧基较近的C原子会与O原子产生相互作用而形成褶皱。Al/GO/Al界面模型中, Al原子与O原子产生相互作用, 二者之间存在离子键作用; C原子与Al原子、O原子之间基本无相互作用。
2)随GO上环氧基数量增加, GO与Al基体之间的界面粘附功提升同时界面能降低。随环氧基增多, Al/GO/Al界面模型的界面结合性和稳定性越强, 复合材料中GO上残留的含氧官能团有利于界面结合。
3)所有含缺陷Al/GO/Al界面模型中, 界面位置O原子与Al原子相互作用不受缺陷影响。不同界面模型中C原子与Al原子之间相互作用程度不同, 单空位缺陷处相互作用最强, 拓扑缺陷处次之, 双空位缺陷处最弱。
4)缺陷位置C原子与Al原子相互作用强于非缺陷位置。两类空位缺陷GO中位于远离环氧基的一侧的缺陷处C原子与Al原子相互作用更强, O原子会抑制空位缺陷处C与Al之间的反应, 保护含空位缺陷GO中碳原子的结构完整性。拓扑缺陷处C原子与界面处环氧基共同与Al原子产生相互作用, 界面结合效果得到提升。
[1] LIU J H, KHAN U, COLEMAN J,. Graphene oxide and graphene nanosheet reinforced aluminium matrix composites: powder synthesis and prepared composite characteristics., 2016, 94(5): 87–94.
[2] GHASALI E, SANGPOUR P, JAM A,. Microwave and spark plasma sintering of carbon nanotube and graphene reinforced aluminum matrix composite., 2018, 18(4): 1042–1054.
[3] ZHANG C X, ZENG Y P, YAO D X,. The improved mechanical properties of Al matrix composites reinforced with oriented-Si3N4whisker., 2019, 35(7): 1345–1353.
[4] ZHANG L, HOU GM, ZHAI W,. Aluminum/graphene composites with enhanced heat-dissipation properties by-reduction of graphene oxide on aluminum particles., 2018, 748: 854–860.
[5] XU Z, BANDO Y, LIU L,. Electrical conductivity, chemistry, and bonding alternations under graphene oxide to graphene transition as revealed byTEM., 2011, 6(5): 4401–4406.
[6] CASTRO-NETO A H, GUINEA F, PERES N M R,. The electronic properties of graphene., 81(1): 109–162.
[7] LEE C, WEI X D, KVSAR J W,. Measurement of the elastic properties and intrinsic strength of monolayer graphene., 2008, 321(5887): 385–388.
[8] CAO M, LUO Y Z, XIE Y Q,. The influence of interface structure on the electrical conductivity of graphene embedded in aluminum matrix., 2019, 6(13): 1900468.
[9] 林启民, 崔建功, 鑫颜, 等. 单点缺陷氧化石墨烯电子结构与光学特性的第一性原理研究. 物理学报, 2020, 35(10): 1117–1122.
[10] KUANG D, HU W B. Research progress of graphene composites., 2013, 28(3): 235–246.
[11] HWANG J, YOON T, JIN S H,. Enhanced mechanical properties of graphene/copper nanocomposites using a molecular-level mixing process., 2013, 25(46): 6724–6729.
[12] ZHANG X, SHI C S, LIU E Z,. Effect of interface structure on the mechanical properties of graphene nanosheets reinforced copper matrix composites., 2018, 10(43): 37586–37601.
[13] ZHANG X, SHI C S, LIU E Z,. Achieving high strength and high ductility in metal matrix composites reinforced with a discontinuous three-dimensional graphene-like network., 2017, 9(33): 11929–11938.
[14] AZAR M H, SADRI B, NEMATI A,. Investigating the microstructure and mechanical properties of aluminum-matrix reinforced-graphene nanosheet composites fabricated by mechanical milling and equal-channel angular pressing., 2019, 9(8): 1070.
[15] WANG J Y, LI Z Q, FAN G L,. Reinforcement with graphene nanosheets in aluminum matrix composites., 2012, 66(8): 594–597.
[16] ZHOU H T, XIONG X Y, LUO F,. The study of thedeposition technique for fabricating., 2021, 70(8): 086201.
[17] GAO X, YUE H Y, GUO E J,. Preparation and tensile properties of homogeneously dispersed graphene reinforced aluminum matrix composites., 2016, 94: 54–60.
[18] JIANG Y Y, TAN Z Q, XU R,. Tailoring the structure and mechanical properties of graphene nanosheet/aluminum compositesby flake powder metallurgy via shift-speed ball milling., 2018, 111: 73–82.
[19] LI Z, GUO Q, LI Z Q,. Enhanced mechanical properties of graphene (reduced graphene oxide)/aluminum composites with a bioinspired nanolaminated structure., 2015, 15(12): 8077–8083.
[20] 何小晶, 原梅妮, 李立州, 等. 石墨烯增强钛基复合材料界面的第一性原理研究. 热加工工艺, 2018, 47(10): 96–100.
[21] CHEN Y T, LIU X H, ZHANG T B,. Interface intrinsic strengthening mechanism on the tensile properties of Al2O3/Al composites., 2019, 169: 109131.
[22] XIE H N, CHEN Y T, ZHANG T B,. Adhesion, bonding and mechanical properties of Mo doped diamond/Al (Cu) interfaces: a first principles study., 2020, 527: 146817.
[23] LIU P, XIE J P, WANG A Q,. First-principles prediction of enhancing graphene/Al interface bonding strength by graphene doping strategy., 2020, 517: 146040.
[24] ZHANG X, WANG S Q. Interfacial strengthening of graphene/ aluminum composites through point defects: a first-principles study., 2021, 11(3): 738–752.
[25] TKACHEV S V, BUSLAEVA E Y, NAUMKIN A V,. Reduced graphene oxide., 2012, 48(8): 796–802.
[26] SHEN J F, HU Y Z, SHI M,. Fast and facile preparation of graphene oxide and reduced graphene oxide nanoplatelets., 2009, 21(15): 3514–3520.
[27] ZHAO Z Y, ZHAO W J, BAI P K,. The interfacial structure of Al/Al4C3in graphene/Al composites prepared by selective laser melting: first-principles and experimental., 2019, 255: 126559.
[28] WU Y H, ZHAN K, Yang Z,. Graphene oxide/Al composites with enhanced mechanical properties fabricated by simple electrostatic interaction and powder metallurgy., 2019, 775: 233–240.
[29] KIM D, NAM S, ROH A,. Effect of interfacial features on the mechanical and electrical properties of rGO/Al composites., 2017, 52(20): 12001–12012.
[30] JU J M, WANG G, SIM K H. Facile synthesis of graphene reinforced Al matrix composites with improved dispersion of graphene and enhanced mechanical properties., 2017, 704: 585–592.
[31] CHEN Y C, LIU Y, ZHOU F,. The interface properties of defective graphene on aluminium: a first-principles calculation., 2021, 188: 110157.
First-principles Study on Interface of Reduced Graphene Oxide Reinforced Aluminum Matrix Composites
SUN Ming, SHAO Puzhen, SUN Kai, HUANG Jianhua, ZHANG Qiang, XIU Ziyang, XIAO Haiying, WU Gaohui
(School of Materials Science and Engineering, Harbin Institute of Technology, Harbin 150001, China)
An “aluminum/graphene oxide/aluminum (Al/GO/Al)” interface model with different carbon/oxygen ratio or with different defects was established. Effects of oxygen-containing functional groups and different defects on the interface of reduced graphene oxide/aluminum composites was studied using first principle method based on density functional theory (DFT). The results show that the epoxy group is better than carbon atom to produce obvious charge interaction with aluminum atom in the interface model of Al/GO/Al. The net charge of oxygen atom is –0.98 e while aluminum atom is 0.46 e, which is conducive to the interfacial bonding between reduced graphene oxide (RGO) and aluminum matrix in composites. When the defects exists, the net charge of carbon atoms at the defects in the Al/GO/Al interface model is in the range of –0.05 e to –0.38 e. Interaction between epoxy group and carbon atoms is weak, while interaction between epoxy group and aluminum atoms is significantly intensified. The existence of epoxy group can inhibit reaction between carbon atom and aluminum atom in the vacancy defects, and protect integrity of carbon structure in RGO with vacancy defects. Therefore, this research may provide theoretical guidance for development of high-performance Al/GO/Al matrix composites.
first principle; reduced graphene oxide/aluminum composite; graphene oxide/aluminum interface model; interface property
1000-324X(2022)06-0651-09
10.15541/jim20210438
TB331
A
2021-07-12;
2021-11-02;
2021-12-16
国家自然科学基金(52071117, 51771063); 黑龙江省杰出青年科学基金(JQ2021E002)
National Natural Science Foundation of China (52071117, 51771063); Heilongjiang Provincial Science Fund for Distinguished Young Scholars (JQ2021E002)
孙铭(1998–), 女, 硕士研究生. E-mail: s1257973295@163.com
SUN Ming (1998–), female, Master candidate. E-mail: s1257973295@163.com
张强, 教授. E-mail: zhang_tsiang@hit.edu.cn; 修子扬, 副教授. E-mail: xiuzy@hit.edu.cn
ZHANG Qiang, professor. E-mail: zhang_tsiang@hit.edu.cn; XIU Ziyang, associate professor. E-mail: xiuzy@hit.edu.cn
