新型牡蛎相关圆环病毒基因组的鉴定
2022-08-23杨李玲郭迎香位红颖方艺菲姜敬哲
杨李玲,郭迎香,位红颖,王 萌,方艺菲,朱 鹏,姜敬哲
1. 天津农学院,天津 300384
2. 中国水产科学研究院南海水产研究所/农业农村部南海渔业资源开发利用重点实验室,广东 广州 510300
3. 广州美格基因科技有限公司,广东 广州 510005
4. 从化区农业农村局,广东 广州 510925
5. 上海美吉生物医药科技有限公司,上海 201321
6. 上海海洋大学,上海 201306
牡蛎属于软体动物门、双壳纲、珍珠贝目,广泛分布于世界各地,其营养价值高,是全球养殖范围最广、产量最高的海水养殖品种。中国作为最大的牡蛎生产国,2019 年牡蛎产量达到 8 259×104t,占世界总产量的85.3% (数据来源:http://www.fao.org)。然而,受环境变化、陆源污染及种质退化等因素影响,牡蛎病害的发生日趋频繁,对其养殖业的健康可持续发展造成负面影响[1]。
1972年Farley等[2]首次报告了牡蛎病毒性疾病病例,证明在高温条件下牡蛎死亡事件与疱疹病毒存在关联,并将这种病毒命名为1型牡蛎疱疹病毒 (Ostreid Herpesvirus-1, OsHV-1)。该病毒在长牡蛎 (Crassostrea gigas) 中具有高致病性,同时对其他多种双壳类也造成严重影响[3-4]。除OsHV-1外,其他报道过的与牡蛎相关的病毒还包括:乳多空病毒科 (Papovaviridae) 病毒,可能导致牡蛎“卵囊炎”,引起卵子和配子细胞肥大;鳃坏死病毒,属虹彩病毒科 (Iridoviridae),可能是导致20世纪60 年代后期葡萄牙牡蛎 (C. angulata) 种群大规模死亡的主要原因;另外,披膜病毒 (Togaviridae)、呼肠孤病毒 (Reoviridae) 和小 RNA 病毒 (Picornavridae) 在贝类宿主中也有多次报道[5]。牡蛎等滤食性贝类在过滤大量海水获取有机颗粒物营养的同时,也会将水体中的其他物质如病毒、污染物等带入体内,并通过外套膜进行浓缩。因此,如果牡蛎养殖水域受到人类活动影响,牡蛎体内可能还会富集诸如诺如病毒 (Norovirus, NV)、甲型肝炎病毒 (Hepatitis A Virus, HAV) 和星状病毒 (Astrovirus, AV) 等引起人类消化道疾病的食源性病毒,但这些病毒并不是牡蛎的病原[6]。病毒基因组是疾病诊断、流行病检测和病原生物学研究的基础。由于无脊椎动物没有能够稳定传代的细胞系,除了OsHV-1和一些食源性病毒外,其他牡蛎相关病毒的鉴定证据主要基于病理学和电子显微镜观察,很少能够拿到基因组序列[7]。因此,基因组数据的匮乏是牡蛎病害研究进展缓慢的一个主要原因。高通量测序和宏病毒组技术的运用,克服了传统病毒学研究对宿主细胞培养的依赖,极大提高了新病毒的发现和鉴定效率。近些年,已在海洋环境、无脊椎动物、低等脊椎动物及人类等相关研究中得到了广泛应用,鉴定到大量的新型病毒[8-11],极大地扩展了人们对病毒世界的认知。
本研究基于前期工作所获得的华南沿海多地养殖牡蛎宏病毒组数据,进一步运用生物信息学工具对其进行深入挖掘,发现并鉴定到5条新型牡蛎相关圆环病毒基因组,为促进牡蛎病毒及贝类病害相关研究提供参考。
1 材料与方法
1.1 数据来源与筛选
课题组前期工作通过对中国华南沿海多地养殖牡蛎进行宏病毒组测序,得到约25亿条测序原始序列 (reads)[12]。对测序数据进行 Fastp (V0.20.0)[13]质控去除低质量和接头序列,用Megahit (V1.2.9)[14-15]将reads组装成重叠群 (contig) 片段,以NCBI非冗余蛋白质数据库为参考,用Diamond[16](V0.9.24.125) 进行比对注释,并用 Megan 6[17]进一步对注释结果进行分类,筛选其中5条基因组完整的、疑似为圆环病毒科 (Circoviridae) 的病毒序列 (Contig ID:ML2-k141_11943、ML2-k141_27933、QZd1-k141_2132、T5S3-k141_267932、ZHd1-k141_220676)进一步深入分析。
1.2 开放阅读框预测与比对
基于 NCBI ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/) 进行开放阅读框 (Open reading frame, ORF) 预测,其中 Minimal ORF length (nt) 选取 75,Genetic code选取 standard,ORF start codon to use 选择 Any sense codon,忽略 nested ORFs。用预测到的ORF进行Blastp比对,其中数据库选择nr蛋白数据库,e为0.000 001。对于有比对结果的ORF,取一致性最高的蛋白用tblastn方法反向比对本研究中病毒的基因组序列,验证5条ORF序列是否完整,并用DNAstar进行翻译,得到最终的蛋白序列。
1.3 基因组进化树的构建及比较基因组分析
将5条病毒全基因组序列提交至VIPTree(V1.9)[18](https://www.genome.jp/viptree/),选择核酸类型为ssDNA,选择宿主病毒类型为Eukaryote,选择基因预测的遗传密码为Eukaryotic,构建基因组进化树。利用MAFFT[19]将5条病毒基因组序列与近源的病毒基因组序列进行比对,并使用TrimAI[20]进一步修剪以去除对齐不明确的区域,利用 MEGA[21]构建 Neighbor-joining Tree,设置 Bootstrap Replications为 1 000,选择 substitution model为 p-distance,设置 Site Coverage Cutoff为50%。
根据与5条病毒复制酶蛋白一致性最高的参考蛋白序列 (YP_006281010.1),计算参考蛋白与5条病毒序列之间的 Amino Acid Identity (AAI)。下载YP_006281010.1对应的基因组序列 (NC_017843.2),利用 Orthologous Average Nucleotide Identity Tool (OAT) 对 NC_017843.2 和 5 条病毒序列计算两两序列的 Average Nucleotide Identity (ANI),得到两两序列的ANI,使用R的ggcorrplot包对数值进行可视化。同时在VIPtree中进行基因组两两比较分析:设置 positioning of each sequence 为auto,colors为 default,ticks of genomes为 shown,vertical dashed lines为 shown,representation of tBLASTx hits为 normal。
1.4 复制酶蛋白进化树分析
因5条病毒复制酶蛋白序列Blatsp比对的结果存在重叠,因此仅以其中1条序列(ML2-k141_11 943)比对结果前50的序列和5条序列一起构建系统进化树,建树过程参考1.3。
1.5 复制酶蛋白结构域分析
在NCBI上下载ARI44308.1[22],用Jalview对ARI44308.1、YP_006281010.1和这5条病毒复制酶蛋白序列进行结构位点分析,使用ClustalW进行比对,下载保存SVG格式文件。用SMART (http://smart.embl-heidelberg.de/) 软件进行结构域预测,同时采用Swiss model进行三维结构预测。
1.6 病毒丰度分析
为了计算每个病毒的相对丰度值,首先合并5 条病毒基因组序列用 salmon (V0.13.1) index[23]命令生成参考基因组数据集,然后再利用salmon quant[23]命令将全部牡蛎病毒组文库的clean reads逐一映射 (Mapping) 到参考基因组上,统计映射上的reads数,根据调整后的FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) 计算公式计算各病毒的相对丰度值。
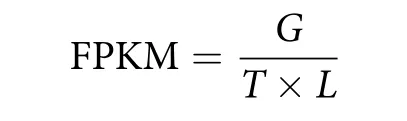
式中:G为比对的reads数;T为测序reads总数(×106);L为基因组长度 (kb)。
2 结果
2.1 病毒注释结果
通过对中国华南沿海多地养殖牡蛎样品进行宏病毒组测序,消除低质量序列并组装,共获得3 375 091 条非冗余 contig序列,进一步与 NCBI的NR库进行blastp比对和分类注释最终获得731 536条疑似病毒来源的contig,筛选其中5条基因组相对完整的、被注释为圆环病毒的基因组序列进一步分析,具体信息见表1。

表1 牡蛎相关圆环病毒基因组信息Table 1 Genome information of oyster-related circoviruses
2.2 基因组水平进化树分析
为了明确病毒的分类,用5条基因组序列与全部单链DNA病毒一起构建基因组水平的进化树(VIPTree,图1)。基因组树中内圈颜色代表病毒科(Family),主要包括圆环病毒科、双生病毒科(Geminiviridae)、类双生病毒科 (Genomovirdae) 和细项圈病毒科 (Anellovridae)。从科水平分类看,圆环病毒科在整个单链DNA基因组树中占据绝大部分。候选的5条序列均聚在了一个小的分支 (红色五角星标识,图1),且在圆环病毒大分支内,说明它们属于圆环病毒科成员。图1的最外圈代表病毒的宿主,包括脊索动物门、节肢动物门、绿藻门和软体动物门。从宿主来源看,候选病毒分支周围主要是节肢动物宿主和其他来源宿主,而节肢动物与软体动物同属无脊椎动物。
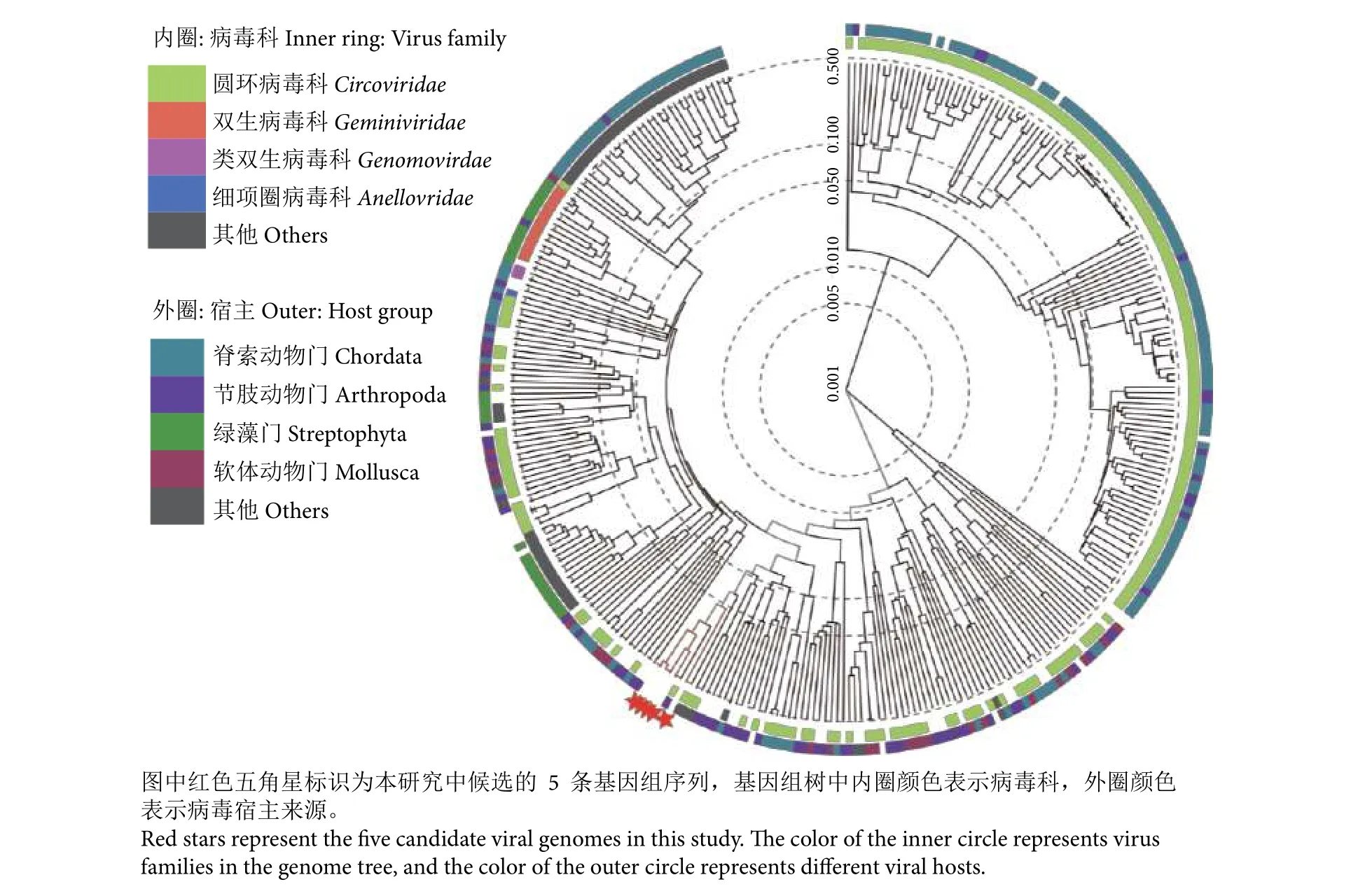
图1 单链DNA病毒基因组进化树 (VIPtree)Fig. 1 Phylogenetic tree of single-stranded DNA virus genome (VIPtree)
为了解5条病毒序列的进化关系,将5条病毒基因组序列与近源的病毒基因组序列进行比对。5 条病毒序列与 NC 017843.2、NC 027797.1、NC 030466.1均聚在了一支,其中除了NC 030468.1来源于海水样品外,NC 017843.2、NC 027797.1、NC 030466.1分别来源于桡足动物、珊瑚、大猩猩样品(图2)。该聚类结果与5条候选序列的来源背景基本一致 (海洋、动物)。值得注意的是,QZd1-k141_2132与T5S3-k141_267932在全基因组水平关系最接近。
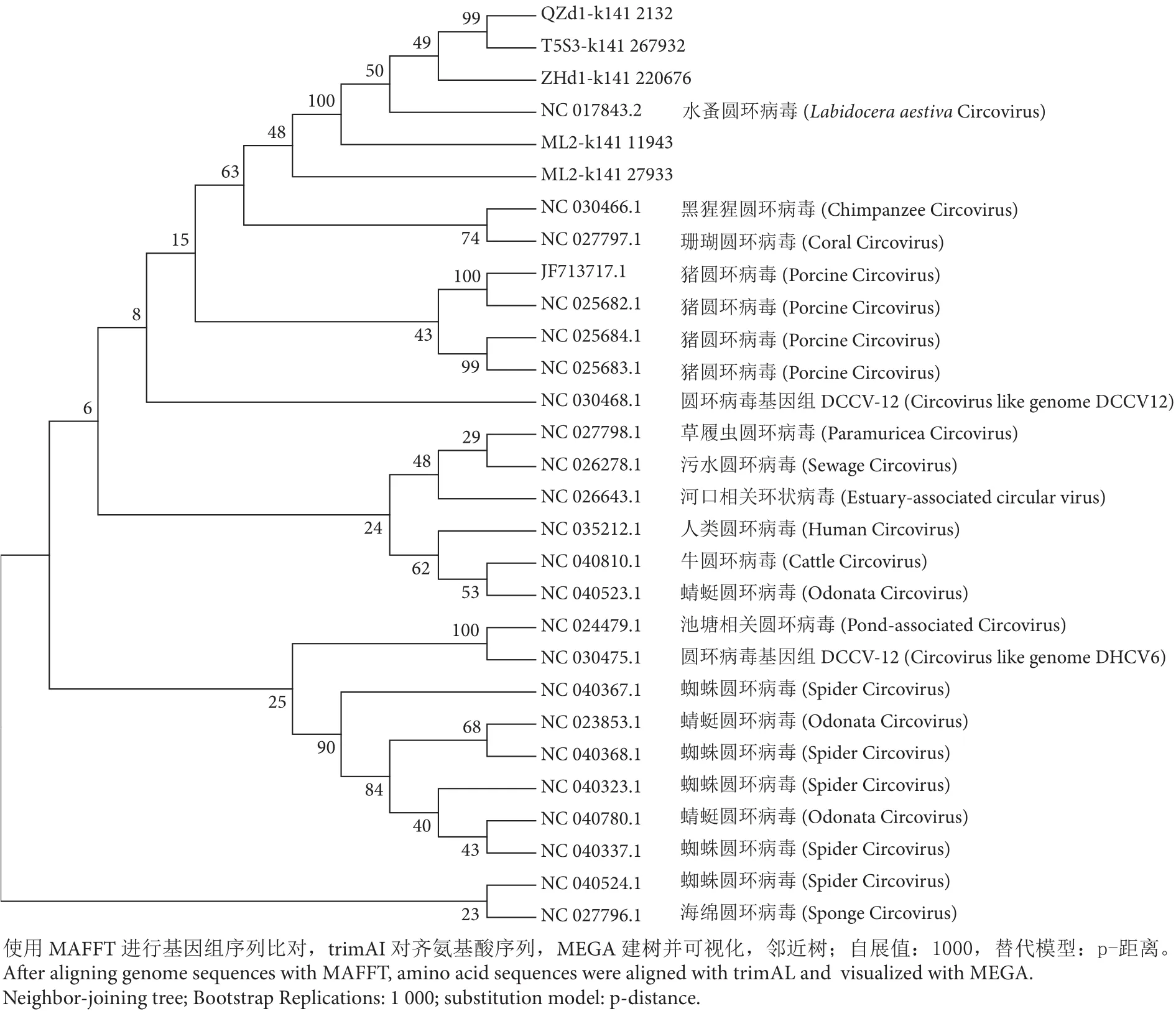
图2 牡蛎相关圆环病毒基因组与近源基因组序列构建的系统发育树Fig. 2 Phylogenetic tree of five oyster-related circoviruses and relative viral genome sequences
2.3 全基因组序列和复制酶蛋白序列一致性分析
用候选序列中预测到的复制酶蛋白序列在NCBI上进行Blastp比对,发现5条病毒序列均与YP_006281010.1的蛋白序列相似性较高。该序列注释为圆环病毒复制酶蛋白,该蛋白来源于一种海洋节肢动物——夏唇角水蚤 (Labidocera aestiva)。用其基因组序列 (NC_017843.2) 和5条序列进行两两比较,NC_017843.2基因组序列与5条候选序列的ANI介于0%~72%,其中最高的是QZd1-k141_2132 (63%),最低的是 ML2-k141_27933 (0%,图3)。5条病毒序列两两间的一致性为0%~94%,其中QZd1-k141_2132与T5S3-k141_267932的最高(94%),ML2-k141_27933和其余4条病毒序列均为0%。根据ICTV规定的圆环病毒科在种水平划分阈值80%来看,5条序列可能属于4个不同的新种,且均与NC_017843.2不同,即QZd1-k141_2132与T5S3-k141_267932可能属于同一种,而其余3条序列属于另外3个种。复制酶蛋白序列一致性分析结果一致。
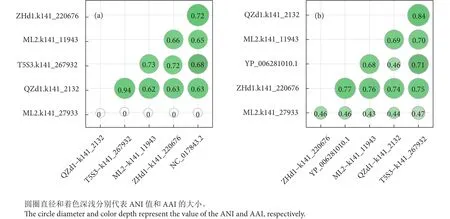
图3 圆环病毒基因组序列间的ANI (a) 和复制酶蛋白序列间的AAI (b)Fig. 3 ANI value among genomic sequences (a) and AAI value among replication proteins of circoviruses (b)
2.4 比较基因组分析
比较基因组分析表明,夏唇角水蚤圆环病毒基因组序列 (NC_017843.2) 与5条候选基因组序列均在复制酶蛋白区域 (即YP_006281010.1) 具有相似性 (图4),但在另一个ORF,即疑似衣壳蛋白区域,均未见明显的相似性。可见候选病毒的衣壳蛋白属于新型的衣壳蛋白,与牡蛎宿主细胞受体的特异性识别相关。
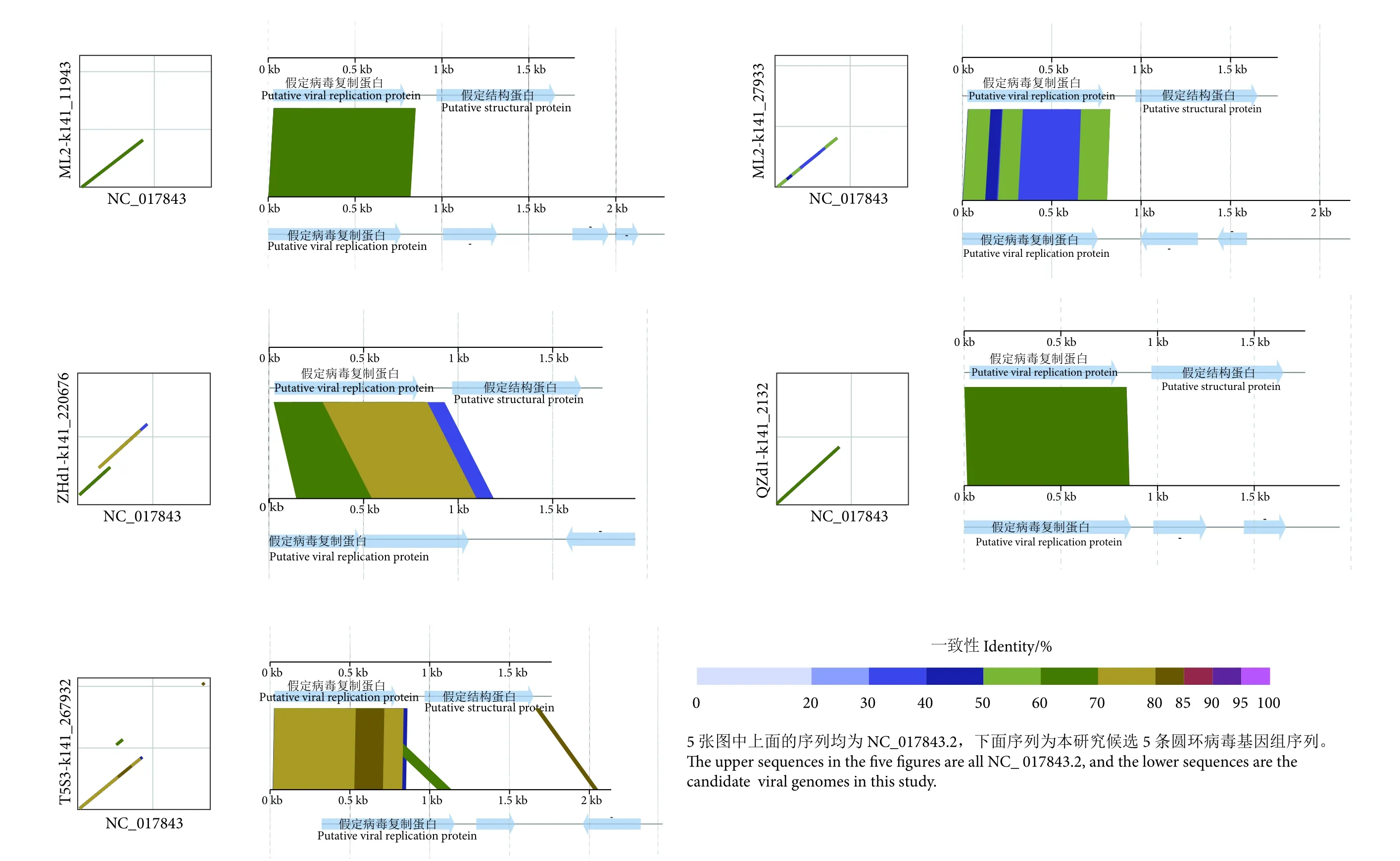
图4 牡蛎相关圆环病毒比较基因组分析图Fig. 4 Comparative genome analysis of oyster-related circoviruses
2.5 Neighbor-joining 进化树构建
因5条病毒复制酶蛋白序列Blatsp比对的结果存在重叠,因此仅以其中1条序列(ML2-k141_11943) 比对结果的前50的序列和5条序列一起构建系统进化树。ML2-k141_11943、ZHd1-k141_220676、QZd1-k141_2132、T5S3-k141_267932 这4个病毒和YP_006281010.1、GAC77785.1、QNG41123.1、AXQ66182.1形成了一个分支,其中YP_006281010.1、QNG41123.1和 AXQ66182.1的来源是动物宏基因组 (具体物种不明),GAC77785.1来源于海洋沉积物 (图5)。ML2-k141_27933序列与AXH76045.1、AXH74760.1形成了一个分支,该序列来源同样是动物宏基因组 (具体物种不明)。

图5 牡蛎相关圆环病毒复制酶蛋白进化树Fig. 5 Phylogenetic tree of replication proteins of oyster-related circoviruses
2.6 功能分析
为查明复制酶ORF中是否有相关功能位点和结构域,用ARI44308.1和YP_006281010.1作为参考序列与5条候选序列进行全局比对 (图6)。虽然5条候选序列在典型环状病毒的复制酶结构域(HUH phage replication sub-family) 处与参考蛋白序列差异较大。但SMART分析发现,除ZHd1-k141_220676外,余下的4条候选序列与2条参考序列都鉴定到明确的HUH结构域。复制酶蛋白结构域三维结构预测表明,5条候选序列,尤其是ML2-k141_11943、ML2-k141_27933和T5S3-k141_267932,与YP_006281010.1复制酶蛋白的空间结构存在较高的相似性 (图7)。

图6 牡蛎相关圆环病毒复制酶蛋白序列比对Fig. 6 Sequence alignment of oyster-related circoviruses
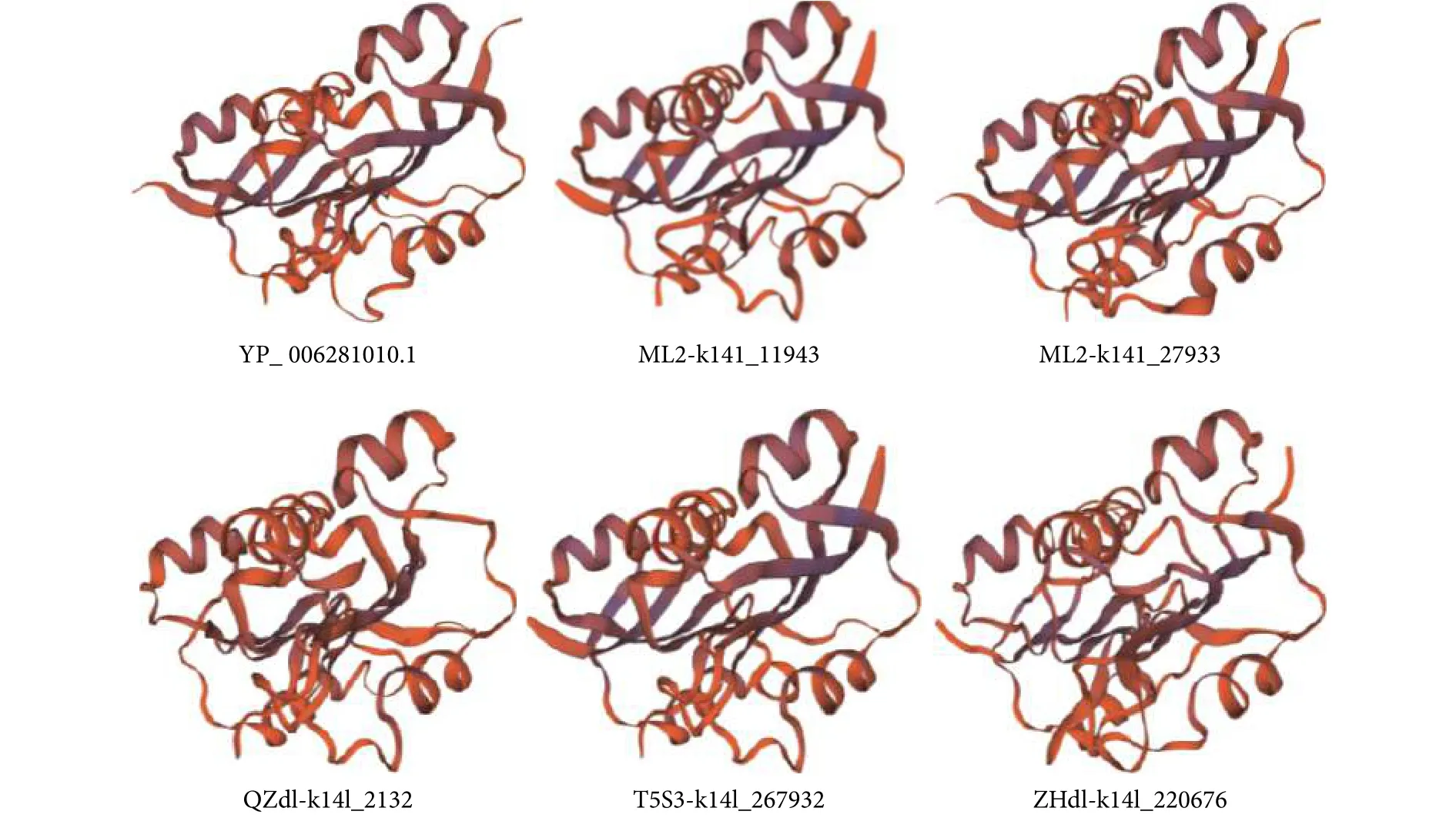
图7 牡蛎相关圆环病毒复制酶蛋白三维结构预测图Fig. 7 Three-dimensional structure prediction of replication protein of oyster-related circoviruses
2.7 病毒丰度分析
为了计算每个病毒的相对丰度值,用salmon统计映射上的reads数。在牡蛎测序文库中均检测到了5条备选病毒序列的reads,FPKM丰度各不相同 (表2)。整体来看,T5S3-k141_267932的分布最广泛,ML2-k141_11943在特定样品中的丰度最高。具体来看,ML2-k141_11943和ML2-k141_27933 在 ML1 中丰度最高 (148.67 和 2 080.33);QZd1-k141_2132和T5S3-k141_267932在ChYJ1509Da中丰度最高 (542.87和76.93);ZHd1-k141_220676在 ChZH1511Da 丰度最高 (55.17)。
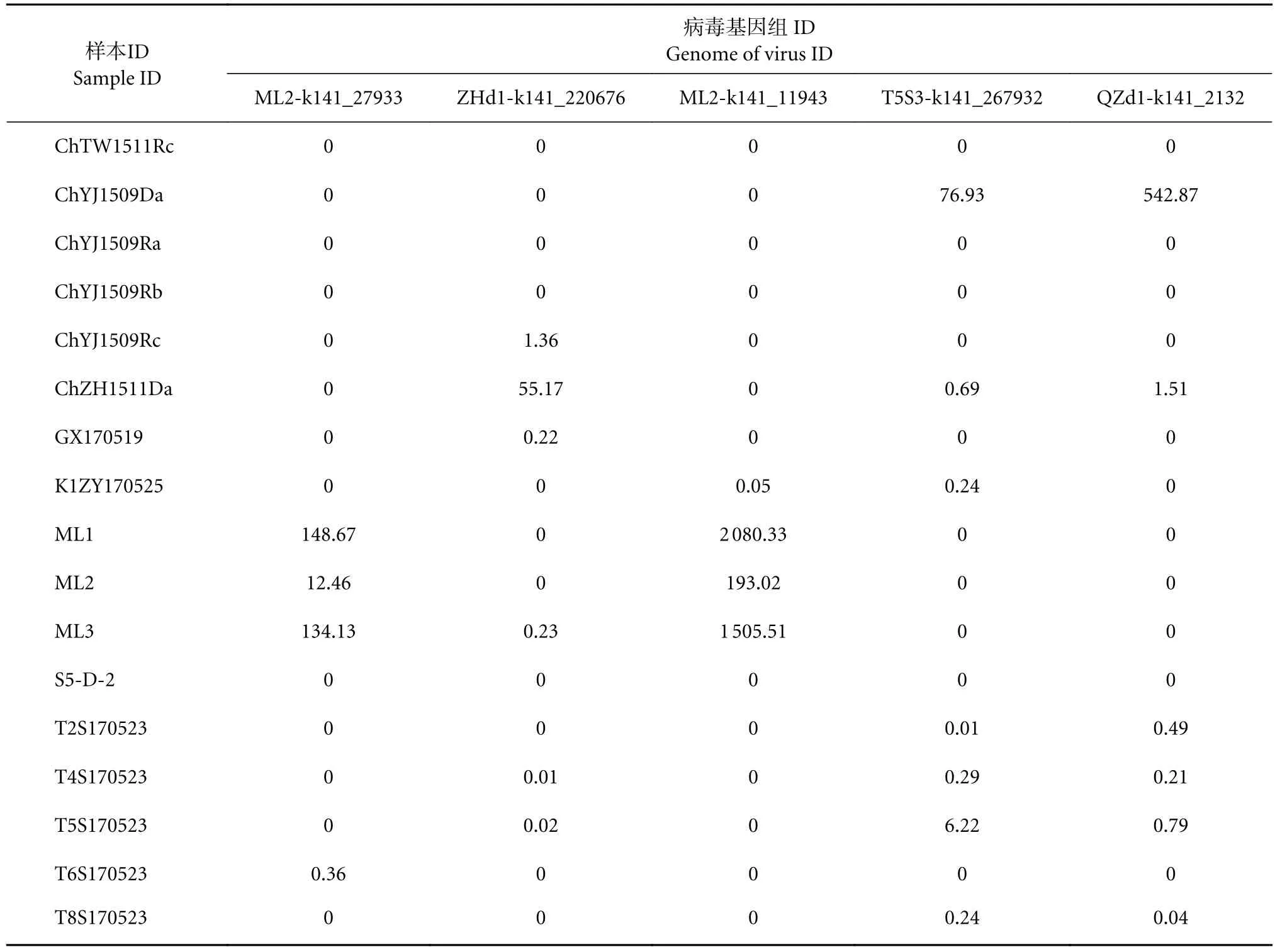
表2 牡蛎相关圆环病毒丰度分析Table 2 Analysis of abundance of oyster-related circovirus virus
3 讨论
近年来,我国沿海地区养殖贝类大规模死亡事件频发,涉及的贝类品种、养殖面积和区域范围不断扩大,使贝类养殖业遭受重大经济损失[24]。传统病毒学研究严重依赖宿主细胞的培养,由于缺乏可稳定传代的无脊椎动物细胞培养技术,加之相关研究投入有限,贝类病毒研究进展十分缓慢[25]。由于高通量测序和病毒组技术的应用,近年几项海洋无脊椎动物病毒研究成果的发表,加深了人们对于海洋动物病毒的了解。2016年,Shi等[26]报道了关于无脊椎动物RNA病毒组的重要研究成果,对包括多种贝类生物在内的约400种无脊椎动物 (包括牡蛎) 组织混合后的约200个文库进行了测序,发现了1 400多种新型RNA病毒,极大地丰富了RNA病毒的多样性和种类。Rosani和Gerdol[27]从长牡蛎 (C. gigas)和地中海贻贝 (Mytilus galloprovincialis) 宿主转录组数据中鉴定了7个几乎完整的RNA病毒基因组,属于小核糖核酸病毒目 (Picornavirales),其中有6种是全新的病毒。Ian等[28]对健康的与被感染的海星进行了比较研究,鉴别出1种细小病毒科 (Parvoviridae) 的疑似致病病原,并且明确其在浮游生物和海洋沉积物中也广泛存在。Cui[29]从不同海域共采集了3门6纲58种的海洋无脊椎动物样品 (包括牡蛎),并利用宏转录组的测序方法对不同物种的RNA病毒组进行研究,鉴定出涵盖9个病毒家族 (Durnavirales、Totiviridae、Bunyavirales、Chuviridae、Picornavirales、Flaviviridae、Hepelivirales、Solemoviridae、Tombusviridae) 的363个RNA病毒,其中的315个与已知的RNA病毒相似度较低,被认为是全新的RNA病毒,其中牡蛎来源病毒4个。牡蛎等海洋无脊椎动物体内可能存在着非常丰富的病毒种类。但前述研究多基于转录组测序数据,发现的新病毒大部分为RNA病毒,对DNA病毒的相关报道则十分有限。
圆环病毒科病毒是已知基因组最小的一类动物致病性DNA病毒,呈二十面体对称球形,无囊膜,直径为17~22 nm,基因组由大约1.7~2.3 kb的单链环状DNA组成。该家族成员种类繁多,可感染各类动物宿主,包括蝙蝠、啮齿动物、畜禽动物和人类等,其中了解比较清楚的是感染畜禽动物和人类的种类,如鸡传染性贫血病毒 (Chicken Infectious anemia)、猪圆环病毒 (Porcine circovirus)、鸽圆环病毒 (Pigeon circovirus)等[30-31]。截止到本研究前,在牡蛎中尚未有圆环病毒的报道。经查询Virus-Host数据库确认 (数据截止至2021年5月),目前已知的所有圆环病毒宿主都是动物界内的两侧对称动物,加之基因组和蛋白序列进化树表明,它们与夏唇角水蚤圆环病毒最为接近,同属无脊椎动物宿主范畴。这意味着这些圆环病毒不可能是牡蛎体内的某些共生细菌或原生动物的病毒。另一方面,圆环病毒的宿主特异性是由其衣壳蛋白序列决定的[32]。而本研究发现5条病毒基因组序列中,仅在ZHd1-k141_220676序列中比对到了疑似衣壳蛋白序列,且与水蚤圆环病毒衣壳蛋白无相似性。因此,这5个新型圆环病毒的宿主是水蚤等节肢动物的可能性不高。当然,现有证据尚不足以确认牡蛎是其真正宿主,还需要结合水体浮游蚤类筛查、扩大养殖牡蛎调查范围及牡蛎人工感染实验来进一步明确其真正的宿主。
根据ICTV标准,同一圆环病毒科物种的成员在其整个基因组中应该具有大于80%的核苷酸同一性[33]。本研究中的5条候选序列与已知圆环病毒在整个基因组水平的相似性低于80%,在衣壳蛋白方面未观察到明显的相似性。因此,本研究鉴定的5个病毒可能与现有圆环病毒科家族成员有较大差异,具体的分类界定还有待发现更多家族成员序列后的分析。
病毒是海洋中最丰富的生命体,以贝类为代表的软体动物门也是海洋中最大的动物类群。但目前对于两者的交叉领域——贝类病毒的了解仍十分有限。宏病毒测序方法已经在众多生物和环境样品中得到广泛应用,突显了高通量测序技术在发现新病毒方面的巨大潜力[12]。然而,病毒组在未知序列解析和新病毒分类方面还有很多不足尚待解决。本研究发现的5个牡蛎圆环病毒的分类地位、家族成员以及宿主范围、致病性、流行情况等方面还有很多工作要做。后续研究应该在发挥病毒组技术优势的基础上,同时配合以经典的病毒学研究方法,获得更为全面和深入的数据,进而为提升为牡蛎等贝类病害的防控水平、支撑相关产业发展发挥作用。
