同时测定酱油中4种甜味剂LC-MS/MS法的建立
2022-08-19雷军
雷军
绵阳师范学院化学与化学工程学院(绵阳 621000)
酱油是我国传统的调味品,因其独特的风味、色泽及丰富的营养价值,已成为我国、日韩、东南亚各国乃至欧美人民饮食生活中不可或缺的调味品[1]。甜菊糖苷和甜菊双糖苷是一类来源于甜叶菊叶片的四环二萜类糖苷物质,被称为继甘蔗、甜菜之后,第三类健康绿色糖源[2-3]。甘草酸和甘草次酸等三萜类化合物是甘草的主要成分,也是甘草甜味的主要来源[4]。近年来,在酱油中陆续检出甜菊糖苷、甜菊双糖苷、甘草酸、甘草次酸等物质。这类物质加入酱油中,可抑制盐味和苦味,增加酱油的风味[5]。但是甜菊糖苷加入酱油中有限量要求、甜菊双糖苷和甘草次酸并未进入食品添加剂管理[6],若在加工过程中为了改善酱油口感超限量、超范围添加此类物质,对人身体有一定的损害。目前,检测其中一种或两种成分的方法有薄层色谱法[7]、高效液相色谱法[8]、液质联用法[9],同时检测这4种组分的方法只有高效液相色谱法[10],这种方法存在灵敏度不高、假阳性以及甜菊糖苷和甜菊双糖苷色谱峰未完全分开的问题[10],导致无法对这4种物质准确定性及甜菊糖苷和甜菊双糖苷准确定量。
针对以上问题,试验建立了同时测定酱油中的甜菊糖苷、甜菊双糖苷、甘草酸、甘草次酸的LC-MS/MS法。在提高此类物质定性能力的同时可以实现甜菊糖苷、甜菊双糖苷完全分离并实现甜菊糖苷和甜菊双糖苷准确定量。
1 材料与方法
1.1 材料与试剂
甲醇(色谱纯,北京百灵威科技有限公司);乙醇(色谱纯,北京百灵威科技有限公司);乙酸乙酯(色谱纯,赛默飞世尔科技有限公司);甲酸(色谱纯,成都市科隆化学品有限公司);无水硫酸镁(分析纯,成都市科隆化学品有限公司);C18粉末(批号:W103678,纳谱分析技术有限公司);甜菊糖苷标准品(天津阿尔塔科技有限公司CAS:57817-89-7 99.7%)、甜菊双糖苷标准品(天津阿尔塔科技有限公司 CAS:41093-60-1 99.8%)、甘草酸标准品(天津阿尔塔科技有限公司 CAS:1405-86-3 80.6%)、甘草次酸标准品(天津阿尔塔科技有限公司 CAS:471-53-4 99.9%)。
1.2 主要仪器与设备
DGU20A-API4500高效液相色谱-串联质谱仪(美国AB SCIEX公司);FA124型电子天平(上海市舜宇恒平科学仪器有限公司);TGL-16型台式高速离心机(四川蜀科仪器有限公司);SHA-B水浴恒温培养振荡器(常州冠军仪器制造有限公司);KQ-700型超声波清洗器(昆山市超声仪器有限公司);ZH-2型旋涡仪(天津药典标准仪器厂)。
1.3 色谱条件
液相色谱柱:ZORBAX Eclipse Plus C184.6 mm×150 mm 3.5 μm;进样体积:10 μL;流速:0.3 mL/min;柱温:40 ℃;流动相:A:甲醇;B:0.1%甲酸。
1.4 质谱条件
DGU20A-API4500液相色谱串联质谱仪,质谱参数:ESI正负离子同时扫描。离子源温度:400 ℃;离子源电压:4 500 V;CUR:20 psi;CAD:5 psi;GS1:30 psi;GS2:30 psi;多反应监测参数如表2所示。
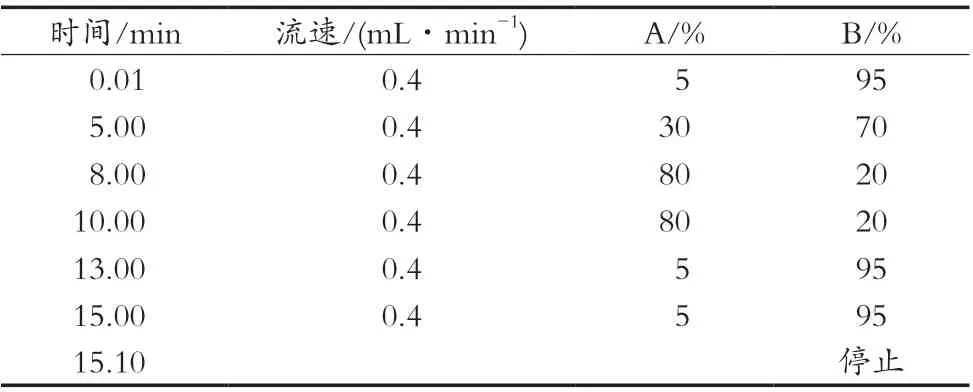
表1 梯度洗脱

表2 多反应监测
1.5 标准曲线的配制
1.5.1 4种甜味剂准储备液的制备
精确称取10 mg甜菊糖苷标准品、甜菊双糖苷标准品、甘草酸标准品、甘草次酸标准品至4个10 mL容量瓶中,用甲醇溶解并定容至刻度,此溶液4种甜味剂的质量浓度为1 mg/mL,冷藏保存。
1.5.2 4种甜味剂标准混合中间液的制备
准确移取甜菊糖苷、甜菊双糖苷、干草酸、甘草次酸标准储备液各0.01 mL至10 mL容量瓶中,用甲醇稀释并定容至刻度,此混合溶液中4种甜味剂的质量浓度为1 μg/mL。
1.5.3 4种甜味剂混合基质标准工作溶液的制备
分别称取6份空白酱油样品各2.0 g(精确至0.01 g)置于25 mL离心管中,准确移取0.0,0.1,0.2,0.4,0.6,0.8和1.0 mL甜菊糖苷、甜菊双糖苷、干草酸、甘草次酸标准混合中间液,按照1.6小节进行提取和净化,配成质量浓度为0,4,8,16,24,32和40 ng/mL的混合基质标准工作液。
1.6 供试品溶液的制备
1.6.1 提取
称取2.0 g(精确至0.01 g)混匀后的酱油试样置于25 mL塑料离心管中,加入10 mL甲醇、4 g无水硫酸镁、2 g C18粉末,旋涡1 min,振荡提取10 min,超声提取10 min,按10 000 r/min离心5 min,取上清液置于另一25 mL塑料离心管,重复上述步骤,合并两次上清液,用甲醇定容至25 mL,待净化。
1.6.2 净化
取1 mL上述待净化溶液至于10 mL塑料离心管中,加入100 mg无水硫酸镁、50 mg C18粉末,涡旋混合2 min,按10 000 r/min离心5 min,过0.22 μm有机相微孔滤膜,滤液供上机分析。
1.7 提取溶剂的选择
根据4种甜味剂的特性,试验依此选择水、乙醇、甲醇、乙酸乙酯为提取溶剂,以质量分数均为100 μg/kg的甜菊糖苷、甜菊双糖苷、干草酸、甘草次酸为质控样,按照1.6小节供试液的制备步骤,比较测得的含量。
1.8 提取次数的选择
采用1.7小节确定的提取溶剂,按照1.6.1小节的提取步骤,以质量分数均为100 μg/kg的甜菊糖苷、甜菊双糖苷、干草酸、甘草次酸为质控样,提取1,2,3和4次,其余步骤同1.6小节,比较测得的含量。
1.9 监测模式的选择
将4种甜味剂标准混合中间液注入液质联用仪,分别记录这4种物质的一级质谱图,分别选取特异性强的监测模式作为试验中甜菊糖苷、甜菊双糖苷、干草酸、甘草次酸的监测模式。
1.10 方法的准确性
称取9份2.0 g混匀后的空白酱油试样于25 mL塑料离心管,3份为一组,分别加入0.1,0.4和0.8 mL甜菊糖苷、甜菊双糖苷、干草酸、甘草次酸标准中间溶液,按照1.6小节供试品溶液制备方法操作,配制成为50,200和400 μg/kg的甜菊糖苷、甜菊双糖苷、干草酸、甘草次酸质控样。
1.11 精密度试验
称取6份2.0 g混匀后的空白酱油试样于25 mL塑料离心管,按照1.6小节样品制备方法,配制成质量分数为100 μg/kg的甜菊糖苷、甜菊双糖苷、干草酸、甘草次酸质控样。将上述6份溶液注入液相色谱-质谱/质谱仪,测得甜菊糖苷、甜菊双糖苷、干草酸、甘草次酸的峰面积,分别代入各自基质标准曲线,计算浓度及SRSD值,即得精密度结果。
2 结果与分析
2.1 标准曲线
由表3可知,4种甜味剂在0~40 ng/mL的质量浓度范围内R均>0.99。表明这4种甜味剂在0~40 ng/mL范围内线性良好。故选用0~40 ng/mL为试验的线性范围。

表3 4种甜味剂的标准曲线
2.2 提取溶剂的选择
由图1可知,甘草次酸在水溶液中溶解度较小,甘草酸在乙醇中难溶,除了甘草次酸,其余三种化合物在乙酸乙酯中的溶解度均较小。这四种化合物在甲醇中均有很好的溶解性。可能的原因是上述溶剂的极性不同,化合物按照“相似相溶”原则,在每种溶液中溶解度不同。综上,选择甲醇为试验的提取溶剂。
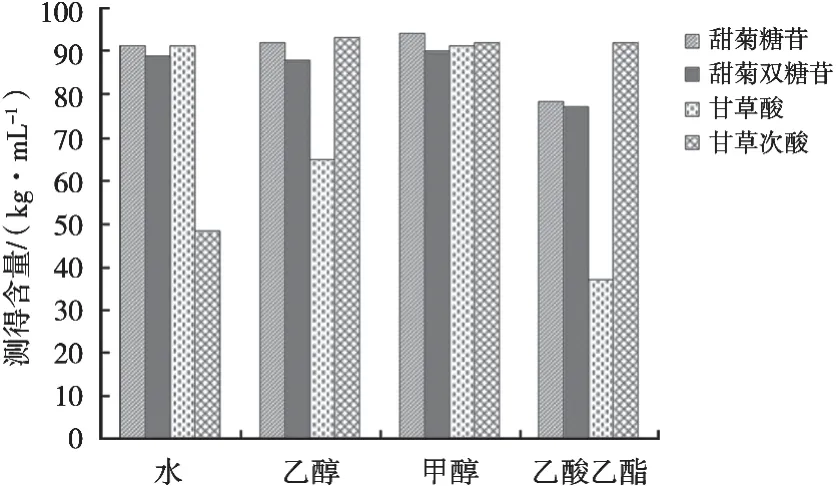
图1 不同提取溶剂的考察
2.3 提取次数的选择
由图2可知,这四种化合物在仅提取一次的情况下,测得值均在80 μg/kg左右,不能提取完全,提取2次以上的测得值明显高于仅提取一次的测得值,且随着提取次数的增加,测得值无明显提升,考虑到操作的简便性、经济性以及环保等因素,试验选择提取2次。

图2 不同提取次数的考察
2.4 监测模式的选择
由图3~图4可知:4种甜味剂在负模式下均有响应,甘草酸在正模式下没有响应;甜菊糖苷和甜菊双糖苷在正模式下以加钠峰的形式出现。由图5~图6可知:甜菊糖苷在负模式下同时出现803.5和641.4两个分子离子峰,严重干扰甜菊双糖苷的分子离子峰,这与朱静雯等[11]研究结果相同。为避免干扰,甜菊糖苷、甜菊双糖苷选取正模式的监测模式;甘草酸、甘草次酸选择负模式的监测模式。

图3 4种物质负模式下的一级质谱图

图4 4种物质正模式下的一级质谱图

图5 甜菊糖苷负模式下的一级质谱图
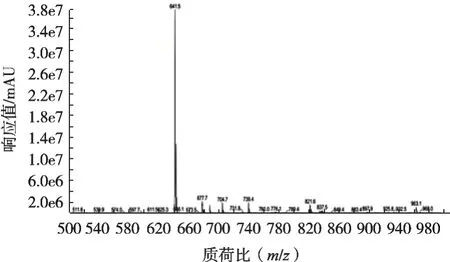
图6 甜菊糖苷负模式下的一级质谱图
2.5 方法的准确性
由表4可知,甜菊糖苷、甜菊双糖苷、甘草酸、甘草次酸在50,200和400 μg/kg回收率依次在90.5%~94.9%,87.7%~94.6%,86.9%~97.3%和87.1%~96.5%之间,满足试验要求。

表4 不同浓度下4种甜味剂回收率结果
2.6 方法的精密度
由表5可知,4种甜味剂用相对标准偏差SRSD表示,SRSD<5%。表明此方法的精密度良好,符合要求。

表5 4种甜味剂精密度结果(n=6)
3 结论
试验建立了同时测定酱油中甜菊糖苷、甜菊双糖苷、甘草酸、甘草次酸的LC-MSMS法,优化了试验的提取溶剂及次数,考察了方法的线性方程、监测模式、专属性、检出限、定量限、方法回收率以及精密度。该方法的监测模式为甜菊糖苷、甜菊双糖苷正模式;甘草酸、甘草次酸为负模式。该方法具有操作简单、定性能力强、灵敏度高等特点,解决了现有液相方法定性能力不强、甜菊糖苷和甜菊双糖苷因无法完全分开导致的定量不准确的问题。
