遗传病注释数据库中基因与变异名称的校验及更正
2022-08-16王雅琼董欣然吴冰冰王慧君卢宇蓝周文浩
王 潇 王雅琼 董欣然 吴冰冰 王慧君 卢宇蓝 周文浩
(国家儿童医学中心/复旦大学附属儿科医院儿科研究院分子医学中心 上海 201102)
近年来,高通量测序技术(全基因组测序、临床外显子组测序以及基因检测包)在遗传病诊断中扮演着重要角色。从测序数据中识别致病遗传变异,能帮助临床医师明确疾病病因,优化疾病管理方案。高通量测序数据分析的主要流程包括对测序数据的预处理及变异检测、变异注释、变异筛选和变异分类等[1]。其中,变异的注释是测序数据分析的基础。简单来说,注释的内容主要包括:数据质量、变异的基因组位置、所属基因及转录本、基因型、人群频率、对mRNA 及蛋白质的影响、致病性预测,以及疾病相关性;在基因层面,还包括基因名、基因功能、表达模式、参与的通路以及相关的疾病或表型等[2]。目前已开发出许多成熟的注释工具如ANNOVAR[3]、VEP 等[4],可 以 对 变 异 进 行 自 动 注释。这些工具依赖的数据库包括公共疾病数据库,如人类孟德尔遗传疾病在线数据库(Online Mendelian Inheritance in Man,OMIM)[5]、人类基因突变数据库(the Human Gene Mutation Database,HGMD)[6]和ClinVar[7]等。OMIM 目前已收集了超过1.6 万个基因和8 600 个表型信息。HGMD 通过人工收集和审核出版文献中的遗传变异信息,截至2020 年6 月收录超过1 万个基因的28 万个与疾病相关的遗传变异。ClinVar 是一个面向公众免费的数据库,一千三百多个机构向其提供了超过80 万条条目,包含超过50 万个与疾病相关的遗传变异及相关注释信息。
在人类基因组学迅猛发展的历程中,多种基因名和基因注释版本并行,同一个遗传变异在基因层面和转录本层面有不一致表示方式,这给临床应用和科研交流造成极大的困扰,甚至会导致疾病诊断失败[8]。目前行业内对于变异的命名主要依据人类基因组变异协会(Human Genome Variation Society,HGVS)标准[9],基因名主要依据人类基因命 名 委 员 会(HUGO Gene Nomenclature Committee,HGNC)提供的核准基因名[10-11]。然而由于疾病数据库中收集的信息来源广、时间长,有的甚至在人类基因组计划开展之前,导致基因及变异的命名方式不符合最新标准。此外,注释所参考的数据库版本也在不断更新。美国国家生物技术信 息 中 心(National Center for Biotechnology Information,NCBI)提供了全面且权威的基因组检索数据库,包含可供检索的基因查询号(Entrez Gene ID)[12]和参考序列查询号(RefSeq ID)[13]。欧洲生物信息所(EMBL-EBI)维护的Ensembl 数据库同样记录了所有基因及参考序列的查询号[14]。GENCODE 是基因组功能注释中最常用的数据库,整合ENSEMBL 的人工和自动基因注释信息,提供对 应 RefSeq 和 ENSEMBL 查 询 号 信 息[15]。GENCODE 从2009 年3 月发布的v2b 开始,平均每2~3 个 月 更 新 一 版(https://www.gencodegenes.org/human/releases. html)。最近一次的更新主要完善新的蛋白质编码基因、lncRNA 以及假基因的注释等[16]。如此高的更新频率也会导致注释数据库中存在一些尚未更新或被弃用的信息。随着大部分注释数据库中的信息数量增长减缓,总体数量已趋于稳定,可以对数据库中的基因及变异名称的准确性进行校验并提供符合指定参考标准版本的正确表示方式。
我们以GENCODE 2020 年4 月发布的版本v34作为参考标准,对OMIM(2020 年7 月版本)中的所有疾病相关基因名及HGMD(2020 年7 月版本)、ClinVar(2020 年7 月版本)中的变异逐一进行比对校验。对校验后有出入的变异提供依据指定注释参考更新后的HGNC 基因名称,转录本查询号(RefSeq,ENSEMBL)和突变表示方式(HGVS)。该工作可以有效提高遗传变异数据分析、解读、验证和交流的效率,辅助遗传病诊断和相关科研工作的顺利进行。
资料和方法
数据来源OMIM 基因注释信息下载自OMIM 网站(https://omim.org/downloads,2020 年7 月);HGMD 变 异 注 释 文 件 下 载 自HGMD 网 站(http://www.hgmd.cf.ac.uk/ac/index.php,2020 年7月);ClinVar 变异注释文件下载自NCBI ClinVar 网站(https://www.ncbi.nlm.nih.gov/clinvar/,2020 年7 月);人基因组转录本注释信息(ENSEMBL 转录本)以及ENSEMBL 转录本查询号与NCBI RefSeq转录本查询号对应列表均下载自GENCODE 网站(https://www. gencodegenes. org/human/release_34lift37.html,版本34,2020 年4 月)。基因注释信息(包含Entrez 及ENSEMBL 基因查询号)同样下载自GENCODE 官 网(版 本19 和34,2020 年4 月)。HGNC 的核准基因名及对应到其他数据库的查询号 信 息 下 载 自HGNC 数 据 库(https://www.genenames.org/download/custom/,2020 年7 月)。所用人基因组版本均为GRCh37。
OMIM 基因名校验由于OMIM 对每个基因提供了OMIM 查询号及其对应的NCBI Entrez 及ENSEMBL 查询号,我们对OMIM 的所有基因分别给出校验后的HGNC 和GENCODE 两种标准基因名,校验结果以参考列表的形式展示(表1)。具体做法如下:(1)以HGNC 为标准的校验。从HGNC网站下载到HGNC 核准基因名与OMIM 查询号、Entrez 基因查询号的对应关系列表。对OMIM 中的所有基因首先按照OMIM 查询号在HGNC 中进行检索,给出其对应的HGNC 核准基因名;若该基因在HGNC 中未匹配到OMIM 查询号,则进一步用OMIM 提供的Entrez 基因查询号在HGNC 中检索并给出对应的HGNC 核准基因名;若仍未匹配到,则认为其没有HGNC 核准基因名,标记为noOMIM2HGNC;若OMIM 未提供某个基因的Entrez 基因查询号,则直接标记为noEntrez。(2)以GENCODE 为标准的校验。在GENCODE v19 和v34 两个版本的数据库中用ENSEBML 查询号进行检索,给出其对应的GENCODE 标准基因名;若该基因在GENCODE v19 或v34 的版本中未匹配到ENSEMBL 查询号,则认为其没有GENCODE 标准基因名,标记为noOMIM2ENSG;若OMIM 未提供某个基因的ENSEMBL 查询号,则直接标记为noENSG。
HGMD 及ClinVar 的变异匹配校验由于HGMD 和ClinVar 中 的 变 异 以HGVS 规 则 展 示,我们以GENECODE 数据库版本v34 中记录的NCBI RefSeq 和ENSEMBL 参考序列查询号为标准,对所有变异以HGVS 规则表示时所使用的参考序列查询号做校验,校验结果以参考列表的形式展示。具体做法如下:(1)查看数据库是否提供变异的HGVS 名称,若未提供相应的HGVS,则在HGVS DNA,HGVS protein,RefSeq 及ENSEMBL 列各标记为无转录本编号(noNM)、无蛋白质编号(noNP)、无 RefSeq 查 询 号(noRefSeq)和 无ENSEMBL 查询号(noENST)。(2)若数据库提供了变异的HGVS,则判断变异是否发生在外显子上,若不在外显子上,进一步判断该变异HGVS 所属RefSeq 转录本是否存在(在GENCODE v34 版本中是否有ENSEMBL 转录本与之匹配),若不存在,则将RefSeq 列和ENSEMBL 列标记为noSite2NM 和noSite2ENST;若RefSeq 转录本存在,则匹配当前版本RefSeq 转录本及ENSEMBL 转录本查询号。若RefSeq 转录本无法匹配到ENSEMBL 转录本,则将ENSEMBL 列标记为noENST2NM。(3)若变异的HGVS 显示其发生在外显子上,同样判断HGVS 中的RefSeq 转录本是否存在。若不存在,则从变异的基因组坐标入手,利用bedtools intersect 工具将其匹配到所有可能的ENSEMBL 转录本上,若没有匹配,则ENSEMBL 列标记为noENST2Site。进一步匹配到当前版本的RefSeq 转录本并给出查询号,若没有匹配,则RefSeq 列标记为noNM2 ENST。(4)若 变 异 处 于 外 显 子 且HGVS 中 的RefSeq 转录本存在,则将该RefSeq 转录本匹配到当前版本的RefSeq 转录本查询号,并匹配到ENSEMBL 转 录 本,若 未 匹 配,ENSEMBL 列 标 记为noENST2NM。若匹配到ENSEMBL 转录本,则查询变异所处的基因组坐标是否位于该ENSEMBL 转录本上。若不在该转录本上,则按照变异的基因组坐标匹配所有可能的ENSEMBL 转录本,若没有匹配,则RefSeq 列和ENSEMBL 列各标记为noNM2Site 和noENST2Site。进一步匹配到当前版本的RefSeq 转录本并给出查询号,若没有,则RefSeq 列标记为noNM2ENST。否则即校验通过。
结 果
OMIM 基因名的校验为了确保在基因诊断和研究交流时采用正确的基因,我们对最常用的遗传病致病基因数据库OMIM 中的所有基因名进行校验。基于NCBI 的Entrez 数据库和ENSEMBL 数据库提供的基因唯一查询号分别比较OMIM 基因与HGNC 和GENCODE 对应的基因名是否吻合(图1A)。校验结果见图2,信息示例见表1,共有17 204 个OMIM 基 因 编 号,对 应17 201 个 基 因,其中有3 个基因是重复的,即TEC,PLCXD1和XGR。在OMIM 更 新 版 本 中(2020 年10 月),TEC和PLCXD1已保留了唯一正确的条目,而XGR(处于X 与Y 染色体同源区域)已被移除。86.7% 的OMIM 给出的基因名与HGNC 的核准基因名是相同的,但仍然存在小部分基因名缺失或错误的情况,其中有972 个(5.65%)OMIM 基因名与HGNC核准基因名不匹配,另外有277 个(1.61%)基因未提 供Entrez 基 因 查 询 号(noEntrez),1 039 个(6.04%)OMIM 基因给出的Entrez 基因查询号没有匹配到HGNC 核准基因名(noOMIM2HGNC)。例如,STRK1(MIM:606799)没有对应的Entrez 基因查询号(noEntrez),DYT13(MIM:607671)没有匹配的HGNC 核准基因名(noOMIM2HGNC)。再如,MEIR1(MIM:616848)校正后的核准基因名为MIER1,属于拼写错误。另一个例子是GLMN(MIM:601749),在OMIM 中给出了GLML,GVM,VMGLOM3 种非正式的基因名。在所有OMIM 校验失败的基因中,我们列出了73 个与HGNC 核准基因名不符的具有表型描述的OMIM 致病基因及其信息(附表2),需要在进行基因注释及诊断报告时予以注意。
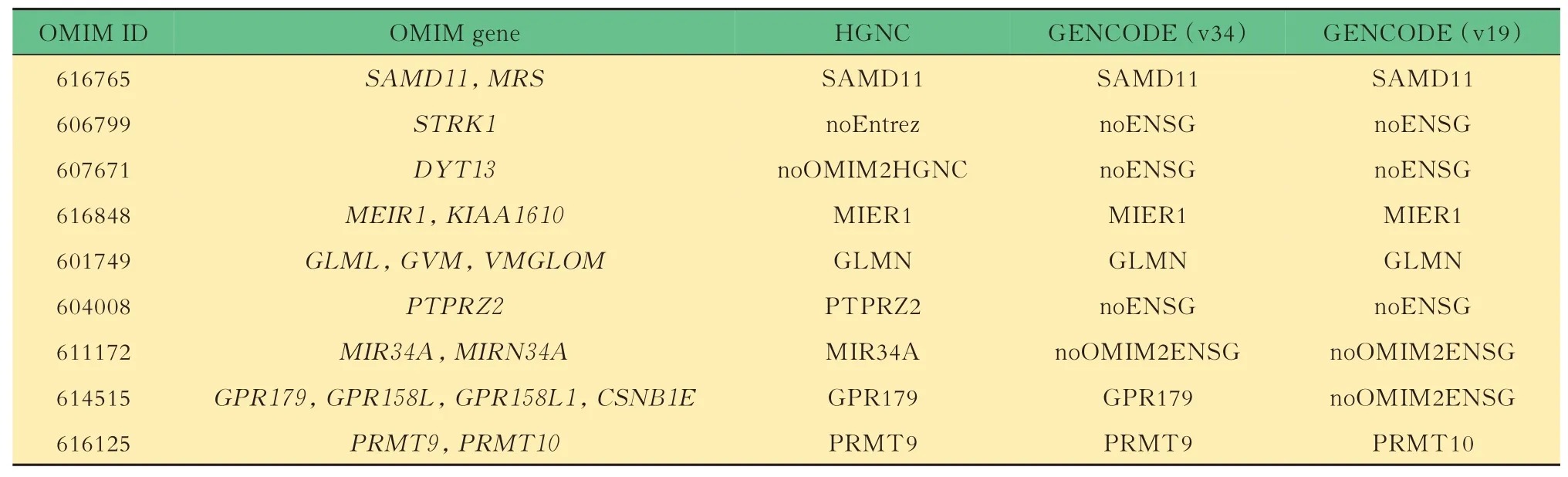
表1 OMIM 基因校验情况示例Tab 1 Examples of gene validation in OMIM

图1 基因名及变异校验方法流程Fig 1 Validation pipeline for gene symbols and variants
OMIM 与GENCODE 数据库比较结果显示1 580 个OMIM 基因没有给出ENSEMBL 的基因查询 号(noENSG,如OMIM 基 因PTPRZ2。 ID:604008),另外有117 个OMIM 基因的ENSEMBLE基因ID 没有匹配到GENCODE 的基因名上(noOMIM2ENSG,如OMIM 基 因MIR34A。ID:611172)。由于基因名本身在不断更新,我们提供了GENCODE 两个版本v19 和v34 的基因名,共有952个OMIM 基因在GENCODE 两个版本中是不同的。 例 如OMIM 基 因GPR179(ID:614515)在GENCODE v19 中 是 缺 失 的;OMIM 基 因PRMT9(ID:616125)在v19 的名字为PRMT10。
HGMD 和ClinVar 变异的校验基因诊断中正确描述致病基因的遗传变异同样至关重要。我们对变异描述遵循HGVS 规则,例如基因区的单核苷酸变异或小片段插入缺失变异需标明参考序列(转录本)、位置和变异类型。由于大量基因对应多个转录本,基因组上同一位置的变异对不同转录本可能造成不同影响,因此确定变异所属的转录本尤为重要。由于预测方法及实验技术的不断更新,转录本本身序列及其查询号都在不断更新,使用错误或滞后的转录本信息会给基因诊断注释及验证带来困扰。因此,对于两个常用的致病变异注释数据库HGMD 和ClinVar,我 们 以GENCODE v34 作 为 参考标准,对HGMD 和ClinVar 提供的每个变异的HGVS 所属转录本进行RefSeq 和ENSEMBL 转录本查询号的匹配和校验(图1B,附表1)。
统计结果见图2、表2。ClinVar 和HGMD 中分别有83.47%和18.78%的变异,与参考注释完全匹配。 对于HGMD,即便忽略蛋白质注释只看mRNA 注释,也仅有21.33%的变异给出的mRNA转录本查询号完全正确。如果从转录本的校验率来看,HGMD 的变异所属的RefSeq 转录本共有10 859 条,仅17.73%与参考注释匹配,ClinVar 的变异所属转录本共有12 291 条,其中98.01%与参考注释匹配。对于HGMD 来说,有78.33%的变异属于其所在的转录本版本不是标准的GENCODE v34的版本(如变异CM1613956,NM_152486.2 校验后应为NM_152486.3),亟待更新。
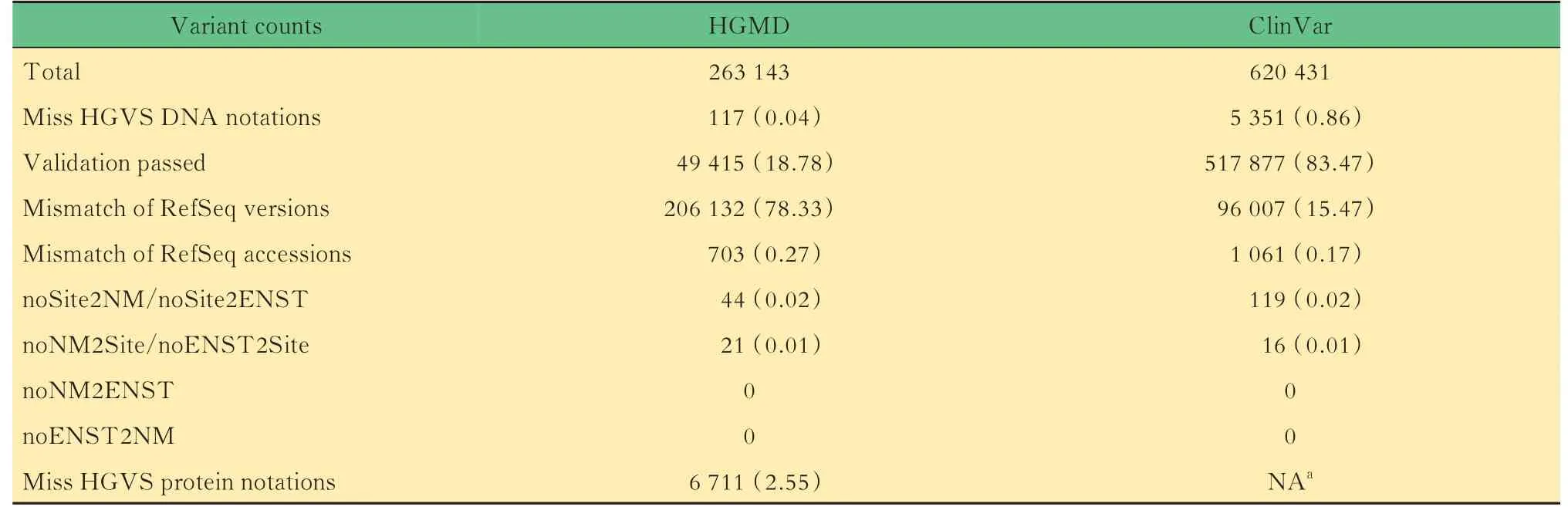
表2 HGMD 和ClinVar 变异校验数量统计Tab 2 Count summary of variants in HGMD and ClinVar [n(%)]

图2 OMIM、HGMD 及ClinVar 校验统计Fig 2 Validation summary of OMIM,HGMD and ClinVar
另外,两个数据库均存在少量变异标注的转录本与参考注释不一致的情况(HGMD:0.27%,ClinVar:0.17%)。如HGMD 变异CD153139 标注转录本查询号为NM_020794.2,该转录本由于缺乏足够的证据而被当前RefSeq 数据库移除(https://www. ncbi. nlm. nih. gov/nuccore/NM_020794.2)。HGMD 数据库中703 个变异共对应47 条独立的RefSeq 转录本(附表3),这些转录本因为缺乏明确的实验证据、不编码蛋白质、包含内含子序列或错误编码到邻近基因等原因已被弃用,或被其他RefSeq 转录本查询号替代(如NM_001257360.1 替代 为 NM_001368809),或 是 NCBI 中 独 有(ENSEMBL 中未找到对应记录)的转录本。
其他校验错误还包括(附表1):HGVS 信息缺失(如CM188806)、非外显子区的变异、HGVS 中的RefSeq 转录本不存在(如HGMD 变异CS1912872所在转录本NM_001291381.1,该转录本通过预测得到,尚未经实验证实)、外显子区的变异HGVS 中的RefSeq 未在GENCODE v34 中收录且根据基因组位置也无法匹配到ENSEMBL 转录本上(如HGMD 变 异 CM1813348 所 在 转 录 本 NM_001171935.1),以及未给出变异所在转录本预测的RefSeq 蛋白质查询号(如HGMD 变异CR133723)。
讨 论
遗传变异的准确表示是变异数据分析的基础,生物信息分析人员常用ANNOVAR、VEP 等综合注释工具对高通量测序分析数据进行一步式注释,而这些工具底层依赖的注释资源是HGMD 等数据库。我们在实际的分子诊断工作中,发现常用疾病数据库注释出来的部分基因或变异的命名是错误的,例如查不到其来源,与文献或其他来源给出的命名不一致,因此对常用注释数据库进行基因和变异的校验是减少注释错误必不可少的一环。我们首次对这些数据库中所有的基因名和变异所属转录本进行名称评估,并建议相关科研及工作人员在实际过程中尽可能选用最新基因注释版本,并在分析报告中标注基因的版本号,便于后续人工核查和追溯。本研究对3 个疾病注释数据库OMIM 基因名和HGMD、ClinVar 的变异进行校验,结果显示数据库中大部分的基因名和变异的注释能与参考注释匹配。然而,在OMIM 中仍有少部分基因存在核准基因名缺失或基因名变更的情况。HGMD 中也存在大量的变异所标注RefSeq 转录本的版本需要更新;HGMD 及ClinVar 中均存在少量变异所标注的转录本已被弃用或查询号改变。在涉及这些基因的变异解读和研究中需要格外注意。
我们选用GENCODE 参考注释,是由于GENCODE 注释系统广泛地应用于大型国际研究项目,如DNA 元件百科全书项目(Encyclopedia of DNA Elements,ENCODE)[17]、基 因 型 和 组 织 表 达关 联 数 据 库(Genotype-Tissue Expression,GTEx)[18]、癌症基因图谱计划(The Cancer Genome Atlas,TCGA)[19]、基 因 组 集 成 联 合(Genome Aggregation Database,gnomAD)[20]、千人基 因组项目(1000 Genomes Project)[21]和人类细胞图谱项目(Human Cell Atlas,HCA)[22]等。采 用GENCODE注释标注便于我们在数据分析过程中整合各大数据库的信息,我们的工具提供新老版本的GENCODE 注释编号,也便于相关人员在变异的解读和后续研究过程中,在各个数据库在线平台进行人工检索查询。由于GENCODE 注释仍在持续更新,本文展现的校验结果具有时效性,但我们开发的方法能较为方便地提供更新的校验结果。除了参考注释,我们所校验的3 个数据库本身也在不断更新与修正。截至2022 年1 月,OMIM 已记录了17 857 个OMIM 条目,与旧版相比,废除了19 个条目,新增672 个条目,在17 185 个同旧版本相同的条目中(OMIM 查询号不变),更新了666 个基因名。新版HGMD(2021 年11 月)收录了315 143 条变异记录,与旧版相比,废除了17 条记录,新增52 017 条记录,对于其中263 126 个查询号未改变的变异,有247 144 个位点的转录本记录发生更新(约94%),进一步证明旧版本的HGMD 中所记录的位点所属转录本号确实存在大量版本滞后的情况。新版ClinVar(2022 年1 月)所记录的变异条目已达到907 441 条,与旧版相比,废除了4 447 条记录,新增291 457 条记录,但是其剩余的615 984 个变异所属的转录本均未在新版本中更新,这也说明我们工作的必要性。特别是在临床报告解读过程中,变异标注的基因名和转录本编号是重要的参考依据,也是后续实验验证的凭据。我们在实践中发现,向实验人员提供准确完整的RefSeq 转录本查询号,包括版本号(如NM_152486.3 中“.3”为版本号)是必要的。因为相同的转录本号下不同的版本代表的mRNA序列有较大差异。
另外,在对变异进行转录本的校验时,我们发现HGMD 中同一个基因组坐标上的变异会有属于多个转录本的情况,这样的变异有18 248 个。在遇到这些变异时,需要人工核查以给出尽量准确的表示方式。在实践中,对于多个转录的情况,一般会优先选择变异影响最严重的转录本,即优先考虑导致无义突变,其次是错义突变的转录本。在基因区域层面上优先考虑位于外显子、剪切位点等区域上的变异所属的转录本,而后考虑位于UTR、内含子或基因间区的转录本。在特定情况下变异影响最严重的转录本不一定是功能最重要的转录本,因此可以采取其他转录本选择方式,例如APPRIS 数据库通过蛋白质结构、序列功能和保守程度为每个蛋白质编码基因定义主要转录本(principal isoform)[23],NCBI 与EMBI-EBI 合作的MANE 项目(Matched Annotation from NCBI and EMBL-EBI,MANE)(https://www. ncbi. nlm. nih. gov/refseq/MANE/)通过专家审核和计算方法整合RefSeq 和ENSEMBL-GENCODE 注释信息,为每个蛋白质编码基因选择高质量的代表性转录组。也有最新研究提出需要综合考虑不同转录本在特定组织中的表达量[24],来选择转录本进行变异的解读。
本文研究了一种便捷的遗传变异表示的校验方法,并提供了更新的注释参考信息,为变异数据分析、临床解读、遗传咨询及科研交流提供了有利的参考依据。
作者贡献声明王潇,王雅琼 数据采集分析,论文撰写和修订,图表绘制。董欣然,吴冰冰,王慧君 可行性分析与监督指导。卢宇蓝,周文浩 论文构思与设计。
利益冲突声明所有作者均声明不存在利益冲突。
