Tumor microenvironment in pancreatic ductal adenocarcinoma:Implications in immunotherapy
2022-08-11CaitlynSmithWeiZhengJixinDongYaohongWangJinpingLaiXiuliLiuFengYin
Caitlyn Smith, Wei Zheng, Jixin Dong, Yaohong Wang, Jinping Lai, Xiuli Liu, Feng Yin
Abstract Pancreatic ductal adenocarcinoma is one of the most aggressive and lethal cancers. Surgical resection is the only curable treatment option, but it is available for only a small fraction of patients at the time of diagnosis. With current therapeutic regimens, the average 5-year survival rate is less than 10% in pancreatic cancer patients. Immunotherapy has emerged as one of the most promising treatment options for multiple solid tumors of advanced stage.However, its clinical efficacy is suboptimal in most clinical trials on pancreatic cancer. Current studies have suggested that the tumor microenvironment is likely the underlying barrier affecting immunotherapy drug efficacy in pancreatic cancer. In this review, we discuss the role of the tumor microenvironment in pancreatic cancer and the latest advances in immunotherapy on pancreatic cancer.
Key Words: Pancreatic ductal adenocarcinoma; Tumor microenvironment; Immunotherapy; Clinical trial;Chemotherapy; Treatment
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) develops in the exocrine compartment of the pancreas and accounts for approximately 90% of pancreatic malignancies, making it the most common pancreatic neoplasm. Due to the lack of early diagnosis and limited treatment response, PDAC remains a highly aggressive and lethal malignancy and is the fourth leading cause of cancer-related death worldwide[1].Although there has been notable progress in understanding tumor biology and the development of novel therapeutic regimens, the average 5-year survival rate is still less than 5%-10% in PDAC patients[1,2]. The clinical manifestations of pancreatic cancers are generally nonspecific, including weight loss,abdominal pain, thromboembolic disease, and type 2 diabetes[3,4]. In approximately 60%-70% of PDAC cases, the tumor arises from the head of the pancreas and could present as pancreatitis and obstructive jaundice[5]. Tumors of the pancreatic body and tail frequently have a poor prognosis due to their late presentation and associated advanced tumor stage[6].
The standard of care for resectable PDAC is surgical resection followed by adjuvant chemotherapy.Surgical resection remains the only curative therapy, but it is available for merely 10%-20% of patients at the time of diagnosis. Moreover, even with curative surgical resection, local recurrence and distal metastasis of PDAC are still quite common[7]. Advanced-stage PDAC is routinely treated with neoadjuvant chemotherapy, and the current first-line therapy regimens include gemcitabine, gemcitabine plus nabpaclitaxel, and FOLFIRINOX (the combination of oxaliplatin, leucovorin, fluorouracil, and irinotecan)[8]. Recently, the poly(adenosine diphosphate-ribose) polymerase inhibitor (PARPi) olaparib (Lynparza)has been approved for patients with germline BRCA-mutated metastatic pancreatic cancer[9]. The development of these neoadjuvant chemotherapy regimens has greatly improved patient survival and quality of life. However, a significant portion of PDAC eventually relapses despite surgical resection and/or neoadjuvant chemotherapy and leads to patient death[10,11].
The difficulties in treating pancreatic cancer lie at the cellular and genetic levels[12]. Mutational changes in pancreatic tumors lead to gene instability, tumor growth, and resistance to treatments[13]. In addition to the characteristic molecular landmarks, including oncogenicKRASmutation and inactivation of the tumor suppressor genesCDKN2A/P16,TP53,andSMAD4, PDAC also frequently harbors mutations involving diverse cell signaling pathways[14]. The molecular heterogeneity likely accounts for its drug resistance in chemotherapy[15]. In addition, pancreatic cancer stem cells,accounting for approximately 1% of all pancreatic cancer cells, have the capacity for self-renewal and exhibit chemoresistance properties[16].
Immunotherapy has emerged as one of the most promising treatment options for advanced solid tumors, including lung, kidney, bladder, liver, and colorectal cancers[17]. Unfortunately, PDAC is notoriously resistant to immunotherapy, and thus far, most phase I/II clinical trials on PDAC have failed to demonstrate the desirable clinical efficacy of immunotherapy[18]. Of note, microsatellite instability (MSI), one of the predictive biomarkers for immune checkpoint blockade therapy, is only detected in a rare small portion of PDAC patients (less than 1%)[19,20]. On the other hand, emerging evidence has pinpointed the tumor microenvironment (TME) in PDAC as a critical component of treatment resistance toward immunotherapy[21,22].
In this review, we discuss the role of the tumor microenvironment and the latest advances in immunotherapy on pancreatic cancer through the search of peer-reviewed clinical and basic research articles related to this topic on PubMed, as well as the publicly accessible information on relevant clinical trials through ClinicalTrials.gov.
TUMOR MICROENVIRONMENT IN PANCREATIC CANCER
PDAC is a type of stromal-rich cancer that frequently presents with a prominent desmoplastic reaction and is characterized by fibrogenic connective stromal tissue surrounding invasive carcinoma[23](Figure 1). Desmoplastic reaction, or desmoplasia, is considered as the morphological basis of the TME.In general, the TME in PDAC demonstrates extensive desmoplasia, decreased stromal vascularization,and altered immune cell infiltration that lead to reduced drug activity and advancement of tumor progression. This process is characterized by an increase in the deposition of noncellular components,such as extracellular matrix (ECM), as well as an increase in the proliferation of cellular components,such as cancer-associated fibroblasts (CAFs) and immune cells[24,25]. Various cytokines, including interferons, interleukins, tumor necrosis factor (TNF), and transforming growth factor β (TGF-β), also play essential roles linking the TME cellular and noncellular components to regulate tumor growth,metastases, and drug resistance. Of note, the overall stroma is responsible for most of the tumor mass,but the stromal cellular components make up a relatively small fraction, approximately 10%-30%, of the tumor mass[26].
Noncellular components of the tumor microenvironment
The ECM is a significant factor in the initiation and progression of PDAC, and its deposition is associated with tumor migration, invasion, and poor prognosis[27]. The ECM is predominantly produced by cancer-associated pancreatic stellate cells (PSCs), a subtype of CAFs[28]. In PDAC, the ECM comprises most of the tumor mass and various matrix proteins, including collagen, fibronectin,proteoglycans, hyaluronan, proteolytic matrix metalloproteinases (MMPs), and tissue inhibitors of MMP[29]. Among ECM components of particular interest are hyaluronan and MMPs in tumor progression and prognosis in PDAC.
In general, ECM provides a rigid barrier leading to increased tumor pressure, decreased vascularization, and reduced drug delivery. A significant cause of drug resistance is the inability of conventional chemotherapeutic drugs such as gemcitabine to penetrate the thick stromal layer[30]. Therefore, it is rational to propose a combinatory therapeutic strategy for PDAC by targeting the tumor ECM.Hyaluronan, or hyaluronic acid (HA), is a glycosaminoglycan polymer and a major component of the ECM. Increased deposition of HA is associated with tumor metastases, drug resistance, and poor prognosis in PDAC[27,31]. Since stromal HA levels are dynamically regulated by synthases (to produce HA) and hyaluronidases (to degrade HA), hyaluronidase-based drug development has been a promising field in targeted therapy against the TME. The enzymatic depletion of hyaluronan through recombinant hyaluronidase (PEGPH20) has led to significantly increased overall survival when combined with neoadjuvant chemotherapy[32]. This is attributed mainly to improved delivery of systemic therapy through degradation of HA and remodeling of the TME. However, a recent phase IB/II randomized study (NCT01959139) of FOLFIRINOX plus pegylated recombinant PEGPH20 showed increased toxicity with this combination therapy and decreased overall survival (OS) (7.7 movs14.4 mo) compared with FOLFIRINOX monotherapy[33]. Moreover, despite promising results of PEGPH20 in phase I-II studies, in a recent phase III randomized study (HALO 109-301), the addition of PEGPH20 to nab-paclitaxel/gemcitabine did not improve OS and progression-free survival (PFS) in patients with hyaluronan-high metastatic PDAC, and additional development of PEHPH20 in metastatic PDAC was halted[34].
MMPs are calcium-dependent metalloproteinases responsible for ECM protein degradation and are implicated in cancer initiation, growth, and metastasis. Clinical trial results with broad-spectrum MMP inhibitors were discouraging due to lack of specificity, associated toxicity, and insufficient clinical benefit[35], warranting further basic and translational studies to classify the role of individual MMPs in PDAC. Among MMP family members, the expression levels of MMP-2, MMP-7, MMP-9, and MMP-11 were significantly elevated in PDAC tumor tissues compared with normal pancreas samples[36,37].Increased MMP-2 expression in PDAC leads to tumor invasion and progression[38-40]. MMP-7 expression is also associated with PDAC initiation and progression[41] and has been shown to be an independent prognostic factor for PDAC in a multivariate analysis. MMP-9 is significantly associated with pancreatic cancer progression and poor prognosis[37] and has emerged as a prognostic biomarker and potential therapeutic target. Highly selective and potent MMP-9 inhibitory antibodies have been developed for ulcerative colitis and colorectal cancer[42]. However, in a preclinical study, systemic ablation of MMP-9 facilitated pancreatic cancer growth and metastasis by creating a tumor-promoting TME[43]. This study has suggested a controversial role for MMP-9 in pancreatic cancer progression.
Additional studies have also demonstrated conflicting results in drugs targeting the tumor stroma.Oliveet al[44] demonstrated that depletion of ECM in PDAC, through inhibition of the Sonic Hedgehog signaling pathway, promoted gemcitabine efficacy and improved survival. However, the involvement of the Sonic Hedgehog-dependent tumor stroma in PDAC has been controversial, as evidence shows that some components of the tumor stroma could actually act to restrain, rather than support, tumor growth[45]. All these failures indicate that targeting desmoplasia alone is insufficient for treating advanced PDAC. The tumor stroma has both tumor-promoting and tumor-suppressing functions,which are probably context dependent. The stromal heterogeneity should be considered for the development of targeted therapy.
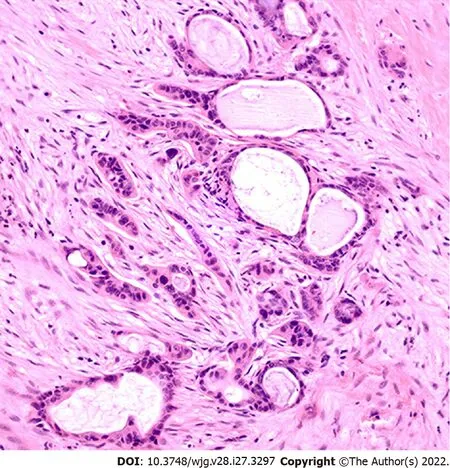
Figure 1 Pancreatic ductal adenocarcinoma with an associated tumor microenvironment. Please note the desmoplastic stromal reaction surrounding the tumor glands, decreased stromal vascularization, and scattered infiltrating inflammatory cells (HE stain, 200 x).
Cellular components of the tumor microenvironment
PDAC displays unique immunologic hallmarks. The TME in PDAC consists of diverse cellular components, including CAFs, regulatory and cytotoxic lymphocytes, macrophages, and endothelial cells[46]. CAFs are the major TME cellular component responsible for the production and deposition of ECM proteins. The involvement of CAFs in the progression of PDAC has been a hot and controversial topic.Similar to the observations made with tumor stroma, CAFs also play dual functions in regulating PDAC progression. On the one hand, CAFs promote cancer progression and drug resistance through the deposition of dense ECM, the release of exosomes (extracellular vesicles), and metabolic support[47-49].On the other hand, depletion of CAFs leads to accelerated PDAC progression and reduced survival in multiple preclinical studies[50,51]. These discrepancies are likely associated with the heterogeneity of CAFs[52,53], a concept supported by recent studies demonstrating the existence of multiple distinct and mutually exclusive CAF subtypes in pancreatic cancer[54,55]. CAF subtypes with diverse biomarkers,including α-smooth muscle actin (αSMA), fibroblast activation protein (FAP), S100A4, and plateletderived growth factor receptor-β (PDGFRβ), have been identified[56]. Specifically, FAP-positive active CAFs have been linked to tumor-promoting functions by maintaining an immunosuppressive TME[57].FAP is a type-II transmembrane serine protease, and its expression has been detected in both the tumor stroma and cancer cells in PDAC, with the highest expression in the tumor stroma at the tumor front[58]. FAP-positive CAFs potently shape the immune landscape in the TME by secreting TGF-β, VEGF,and multiple matrix processing enzymes[59,60], recruiting circulating myeloid-derived suppressor cells(MDSCs) into the tumor stroma[57], and inhibiting natural killer cell (NK) cytotoxicity and cytokine production[61]. FAP has been suggested as an ideal target for the TME, and its specific therapeutic reagents are in development[62].
In addition to CAFs, the TME also consists of multiple types of immunosuppressive cells, including regulatory T cells (Tregs), MDSCs, and tumor-associated macrophages (TAMs)[63]. These cells correlate to provide an immunosuppressive TME and have been under extensive preclinical and clinical investigation.
Tregs, defined as CD4+/CD25+/FOXP3+ T cells, are a subtype of repressive T cells that play an essential role in maintaining immune tolerance and preventing autoimmune disorders. Tregs can be found in PDAC and premalignant lesion intraductal papillary mucinous neoplasms (IPMNs). The prevalence of Tregs in CD4+ T lymphocytes correlates significantly with the progression and invasion of IPMNs and is associated with poor prognosis in PDAC. The immunosuppressive function of Tregs has been attributed to the secretion of suppressive cytokines, including IL-10 and TGF-β1, and the induction of CD4+ T-cell death[64,65]. Preoperative chemoradiation therapy has been shown to decrease Tregs in PDAC[66]. However, in a recent study, depletion of Tregs in a mouse model caused accelerated tumor progression due to unexpected crosstalk between Tregs and CAFs in PDAC[67]. This study has challenged the current view and posed uncertainties in developing Treg-based targeted therapy.
MDSCs and TAMs have also been suggested as potential therapeutic targets against the TME. Even though these two cell types are considered as separate entities, they have no demarcated boundaries and share many common characteristics[68]. MDSCs are a group of heterogeneous immature myeloid cells and can potently suppress T-cell function in tumors[69]. The levels of MDSCs correlate with the progression of PDAC and have been proposed as a predictive biomarker of chemotherapy failure[70,71]. TAMs are circulating monocyte-derived macrophages in the tumor stroma and represent a significant population of immune cells within the TME. TAMs can be further subclassified into the M1 and M2 subtypes, with M1 being proinflammatory (antitumorigenic) and M2 being anti-inflammatory(protumorigenic)[72]. M2-polarized TAMs are associated with an unfavorable prognosis in PDAC[73].Liuet al[74] revealed progressive accumulations of MDSCs and M2-polarized TAMs accompanied by dynamic reductions in cytotoxic T cells (CTLs) and helper T cells (Ths) in PDAC progression.Gemcitabine affects the TME by inhibiting the expansion of MDSCs and the induction of Th2 cells while promoting M2-polarized TAMs[74]. M2-polarized TAMs can also be induced by other chemotherapeutic agents, such as carboplatin and cisplatin, leading to increased secretion of interleukin-6 (IL-6), IL-10, and prostaglandin E2[75]. In addition, interferon-γ upregulates the expression of programmed death-ligand 1 (PD-L1) in MDSCs, resulting in an immunosuppressive environment[76]. Further investigations and clinical trials are needed to test the efficacy of targeting MDSCs and TAMs in pancreatic cancer.
IMMUNOTHERAPY IN PANCREATIC CANCER
Current treatment options for PDAC have limited effects on patient survival. The recent development of immunotherapy has improved clinical outcomes for various types of solid tumors[17] and can revolutionize cancer treatment in PDAC. Activating the patient's T cells is the principal basis for cancer immunotherapy. This is accomplished through multiple mechanisms, such as decreased tumor-specific antigen presentation, T-cell activation, T-cell infiltration into the pancreatic tumor, and elimination of cancer cells by T cells[77]. Multiple cancer immunotherapies have been introduced, including immune checkpoint inhibitors, cancer vaccines, and adoptive cell transfer.
Immune checkpoint inhibitors
Immune checkpoint molecules are a group of surface receptors expressed on various immune cells that transduce inhibitory signals to T cells upon ligand binding. These molecules play an important role in preventing an autoimmune attack against self-antigens. Due to strong immune selective pressure,cancer cells frequently adopt the power of immune checkpoint molecules to avoid immune destruction.Initially approved for the treatment of metastatic melanoma, immune checkpoint inhibitors (ICIs) have been cleared to treat various solid tumors, including advanced or metastatic urothelial carcinoma, nonsmall-cell lung cancer, colorectal cancer, triple-negative breast cancer, and head and neck squamous cell carcinoma[78,79]. Currently, FDA-approved immune checkpoint inhibitors (ICIs) include anti-CTLA-4 agents (ipilimumab), anti-PD-1 agents (nivolumab, pembrolizumab, cemiplimab) and anti-PD-L1 agents(atezolizumab, avelumab, durvalumab)[79].
ICIs have emerged as a new therapeutic option for pancreatic cancer. Unfortunately, most phase I and II clinical trials on ICI treatment have failed to show the desired beneficial effect in PDAC. Two independent phase II clinical trials have demonstrated unsatisfactory clinical outcomes on monotherapy with anti-CTLA-4 mAb (Table 1). Single-agent ipilimumab, an anti-CTLA-4 mAb, was ineffective for the treatment of advanced PDAC (NCT00112580) (https://clinicaltrials.gov/ct2/show/NCT00112580)[80,81]. Monotherapy with tremelimumab, another anti-CTLA-4 mAb, also yielded poor clinical outcomes in PDAC, with 18 out of 20 patients demonstrating progressive disease and a poor median OS of 4 mo(95%CI: 2.83-5.42 mo) (NCT02527434) (https://clinicaltrials.gov/ct2/show/NCT02527434).
Combination therapy with ipilimumab and gemcitabine, on the other hand, has demonstrated promising results due to the increased immune response by enhancing naïve T-cell activation[82]. In a phase 1b clinical trial (NCT01473940) (https://clinicaltrials.gov/ct2/show/NCT01473940), initial results on combination therapy with ipilimumab and gemcitabine showed that the treatment was tolerable, with a median PFS of 2.5 mo (95%CI: 0.8-4.8 mo) and a median OS of 8.5 mo (95%CI: 2.2-10.3 mo). In this study, five out of the 11 patients had stable disease, while two had a partial response. An ongoing clinical trial (NCT01928394) (https://clinicaltrials.gov/ct2/show/NCT01928394) is comparing nivolumab (anti-PD-1 mAb) monotherapy and combination therapy with nivolumab plus ipilimumab in patients with advanced or metastatic PDAC, and the results will be released in 2023.

Table 1 Complete immune checkpoint inhibitor-based clinical trials in pancreatic ductal adenocarcinoma
Notably, in a phase I clinical trial (NCT00556023) (https://clinicaltrials.gov/ct2/show/NCT00556023), a tolerable and safe profile was demonstrated by combination therapy with tremelimumab plus gemcitabine, warranting further study in patients with metastatic PDAC. Thirty-four patients were enrolled in the study, and the median OS was 7.4 mo (95%CI: 5.8-9.4 mo). Two patients achieved a partial response at the end of treatment[83]. A phase Ib/II study (NCT02331251) (https://clinicaltrials.gov/ct2/show/NCT02331251) was performed to evaluate the safety and efficacy of pembrolizumab, an anti-PD-1 mAb, in combination with gemcitabine plus nab-paclitaxel chemotherapy. The median PFS and OS were 9.1 and 15.0 mo for chemotherapy naïve-treated patients,respectively, and changes in tumor cell-free DNA copy number instability were considered to be a potential prognostic factor for OS[84].
A phase I study on atezolizumab, an engineered IgG1 mAb targeting PD-L1, showed tolerability at doses up to 20 mg/kg every three weeks in a Japanese cohort[85]. In a phase II randomized clinical trial(NCT02558894) (https://clinicaltrials.gov/ct2/show/NCT02558894), evaluation of durvalumab, an anti-PD-L1 agent, with or without tremelimumab in patients with metastatic PDAC was evaluated following the failure of 5-FU and gemcitabine-based chemotherapy[86]. No patients in the study responded to durvalumab monotherapy, and the efficacy analysis demonstrated an objective response rate (ORR) of 3.1% (95%CI: 0.08-16.22) with the combination therapy of durvalumab plus tremelimumab[86].
A high tumor mutational burden (TMB) in cancer cells tends to produce more immunogenic neoantigens and may predict immunotherapy response[87]. A phase II clinical trial (NCT05093231) (https://clinicaltrials.gov/ct2/show/NCT05093231) investigating the efficacy of pembrolizumab plus olaparib in metastatic pancreatic adenocarcinoma patients exhibiting high tumor mutation burden is ongoing, and the results will be released in 2026.
Based on the results from current clinical trials, further studies need to focus on the combined approaches using ICIs with different therapeutic approaches, including chemotherapy, radiotherapy, or additional innovative platforms of immunotherapy, such as cancer vaccine and adoptive cell transfer.
Therapeutic cancer vaccines
Therapeutic cancer vaccines include whole-cell vaccines, dendritic cells, DNA, and peptide vaccines that activate cancer antigen-specific cytotoxic T lymphocytes (CTLs), eliciting immunogenic antigen presentation and leading to an anticancer response[88]. One such pancreatic cancer vaccine is GVAX,which is generated from irradiated pancreatic cancer cells expressing granulocyte-macrophage colonystimulating factor (GM-CSF)[89] (Table 2). Upon vaccination, GVAX secretes GM-CSF, induces subsequent activation of antigen-presenting cell and T-cell priming, and stimulates the patient’s immune system against pancreatic cancer cells[90]. GVAX was tolerable even at high doses, and the vaccination-induced increased delayed-type hypersensitivity response to autologous tumor cells[91]. In a phase II clinical trial on GVAX (NCT00084383) (https://clinicaltrials.gov/ct2/show/NCT00084383),sixty patients received GVAX 8-10 wk after surgical intervention, followed by adjuvant 5-FU-based chemoradiotherapy. The median PFS was 17.3 mo (95%CI: 14.6-22.8 mo), with a median OS of 24.8 mo(95%CI: 21.2-31.6 mo), which compares favorably with published data for resected PDAC[92].Combinatory immunotherapy has aimed to induce a much more sustained antitumor T-cell response[93]. In a phase Ib trial for locally advanced, unresectable or metastatic PDAC (NCT00836407) (https://clinicaltrials.gov/ct2/show/NCT00836407), thirty patients received either ipilimumab monotherapy or ipilimumab plus GVAX cancer vaccine, and the median OS was 3.6 mo for the ipilimumab monotherapy group, compared to 5.7 mo in the group with combination therapy[94].Although combinatory immunotherapy has shown its potential for advanced PDAC, more studies are needed to fully explore this novel therapeutic strategy’s capability.
Few human leukocyte antigen (HLA)-A(*)2402-restricted tumor-associated antigens, including the KIF20A-10-66 peptide, have been identified in PDAC[95]. A phase I/II clinical trial in Japan showed a better prognosis in patients with metastatic PDAC and HLA-A*2402-positive status who received KIF20A-10-66 peptide vaccination as second-line treatment after failure of gemcitabine chemotherapy[96]. In two separate phase II clinical trials, KIF20A-derived peptide was evaluated in combination with two antiangiogenic cancer vaccines targeting vascular endothelial growth factor receptor 1 (VEGFR1)and VEGFR2. In the HLA-A*2402-matched group, patients with peptide-specific CTL induction had improved prognosis and increased OS[97,98]. Another HLA-A24-restricted antigenic peptide, SVN-2B,also functions as an immunogenic molecule. A vaccination protocol of SVN-2B in combination with interferon-α has demonstrated effective clinical and immunological responses for advanced PDAC[99].
Algenpantucel-L is a whole-cell pancreatic cancer vaccine with two irradiated allogenic human pancreatic cell lines (HAPa-1 and HAPa-2) expressing the murine enzyme (1,3)-galactosyltransferase (α GT)[100]. Of note, the αGT enzyme is the critical barrier to xenotransplantation due to hyperacute rejection[101]. As a result, Algenpantucel-L will induce a hyperacute rejection of the allograft cells through rapid activation of antibody-dependent cell-mediated cytotoxicity (ADCC), leading to a response against the patient’s pancreatic cancer cells through epitope spreading[102]. A phase II, openlabel trial (NCT00569387) (https://clinicaltrials.gov/ct2/show/NCT00569387) evaluated the use of the Algenpantucel-L tumor vaccine in combination with gemcitabine plus 5-FU chemoradiotherapy in patients with resected PDAC. Seventy patients were recruited in the study, and the 12-mo disease-free survival (DFS) and OS were 63% and 86%, respectively, suggesting that the Algenpantucel-L tumor vaccine could be administered with standard chemotherapy following surgical resection of pancreatic cancer[101]. Unfortunately, in a recent phase 3, open-label, randomized clinical trial (NCT01836432) (https://clinicaltrials.gov/ct2/show/NCT01836432), Algenpantucel-L failed to improve survival on borderline resectable or locally advanced PDAC receiving neoadjuvant chemoradiation therapy[103].
Overexpression of Mucin 1 (MUC-1), a type I transmembrane protein with O-linked glycosylation,plays a crucial role in oncogenic signaling to promote metastasis, angiogenesis, and invasion[104].MUC-1 has served as a target for cancer vaccine immunotherapy[105]. Following surgical resection, a phase I/II study of a MUC1 peptide-loaded dendritic cell vaccine was conducted in 12 pancreaticobiliary cancer patients. Four out of twelve (33.3%) patients who received this MUC-1-based tumor vaccine were alive after four years without evidence of recurrence[106]. An optimized construct with MUC-1-variable number tandem repeats has been designed with much more potent immunogenicity[107].
Dendritic cell vaccines have been introduced to enhance the antitumor immune response through the stimulation of naïve T cells[108]. In a study evaluating the effectiveness of a dendritic cell vaccine in patients with advanced PDAC (NCT01410968) (https://clinicaltrials.gov/ct2/show/NCT01410968),autologous dendritic cells were isolated in HLA-A2-positive patients, loaded with three A-2 restricted peptides, and readministered as a cellular vaccine. The results were promising with the generation of antigen-specific T cells in three patients, as well as tolerable adverse effects[109]. In a phase I study in Japan, a Wilms' tumor 1 (WT1)-pulsed dendritic cell vaccine combined with chemotherapy showed safety and potential acquisition of immunity in resected PDAC[110]. Multiple associated studies have further supported the clinical benefits of dendritic cell-based vaccines in PDAC[111-113].
Approximately 95% of PDAC patients have mutations in theKRASoncogene. Despite an early study suggesting an unproven efficacy by targeting mutated KRAS in PDAC[114], multiple subsequent clinical studies have demonstrated the clinical potential for such a therapeutic approach. A phase I/II clinical trial (NCT02261714) (https://clinicaltrials.gov/ct2/show/NCT02261714) evaluated the efficacy of a synthetic mutant RAS peptide vaccine with GM-CSF in PDAC. TG01, a mixture of 7 synthetic RAS peptides representing the most commonKRASmutations, combined with GM-CSF and gemcitabine was well tolerated with a robust immune response and improved clinical outcome[115]. One study demonstrated a long-term immune response and improved survival in patients with resected PDAC after KRAS vaccination[116]. An alternative KRAS-based tumor vaccine is GI-4000, a recombinant heat-inactivated Saccharomyces cerevisiae yeast-derived vaccine expressing mutated KRAS proteins. A phase I trial revealed a favorable safety profile and immunogenicity of the GI-4000 cancer vaccine[117].A subsequent phase II trial (NCT00300950) (https://clinicaltrials.gov/ct2/show/NCT00300950)compared GI-4000 plus gemcitabine with placebo plus gemcitabine alone in patients with resected PDAC carryingKRASmutation. GI-4000 was well tolerated. It led to a similar median OS compared with placebo. However, compared with the placebo group, the GI-4000 group had a trend of improved OS (523.5vs443.5 d) and an increased frequency of immune responders (40%vs8%) in the stratified R1 resection subgroup[118].

Table 2 Complete vaccine immunotherapy-based clinical trials in pancreatic ductal adenocarcinoma
The GV1001 tumor vaccine consists of a fragment (16 amino acids) of human telomerase reverse transcriptase (hTERT) found in a high proportion in PDAC cancer cells and has been introduced as a novel therapeutic regimen[119]. In a phase I/II clinical trial evaluating the clinical outcomes in patients with unresectable PDAC, GV1001 plus GM-CSF elicited an immune response in 63% of patients,resulting in a median OS of 7.2 mo for immune responders compared to 2.9 mo for nonimmune responders[120]. However, in a randomized phase III study of patients with locally advanced and metastatic PDAC, combination therapy consisting of GV1001, gemcitabine, and capecitabine chemotherapy showed no improvement in OS compared to chemotherapy alone [6.9 mo (95%CI: 6.4–7.6 mo)vs7.9 mo (95%CI: 7.1–8.8 mo)] (NCT00425360) (https://clinicaltrials.gov/ct2/show/NCT00425360)[121]. Another GV1001-based phase III clinical trial (NCT00358566) (https://clinicaltrials.gov/ct2/show/NCT00358566) was terminated early because of a lack of survival advantage.
Adoptive cell transfer
Adoptive cell transfer, also known as cellular immunotherapy, includes chimeric antigen receptor T-cell(CAR T cell) therapy and tumor-infiltrating lymphocyte (TIL) therapy[122,123]. CAR T-cell therapy is the most common type of adoptive cell transfer. Generally, it involves harvesting the patient’s T cells,genetic modification to express surface chimeric antigen receptor,ex vivoexpansion, and then transferring the cells back to enhance tumor immunity. In forty-three patients with PDAC who underwent radical pancreatectomy, gemcitabine plus adoptive cell transfer with T cells stimulated by the MUC1-expressing human pancreatic cancer cell line demonstrated a median OS of 14.7 mo[124].Mesothelin is a tumor antigen highly expressed in PDAC[125]. In a preclinical study, CAR T-cell therapy targeting mesothelin demonstrated promising tumor-suppressive effects[126]. Amatuximab(MORab-009), a chimeric mAb targeting mesothelin, also led to reduced growth of mesothelinexpressing tumors, including PDAC[127]. In a phase I trial, the efficacy of MORAb-009 was tested in seven PDAC patients, and one patient had disease control for greater than six months[128]. However, in a phase II randomized placebo-controlled clinical trial (NCT00570713) (https://clinicaltrials.gov/show/NCT00570713) evaluating the efficacy of MORAb-009 plus gemcitabine, no significantly improved clinical outcome was observed [median OS: 6.5 mo (95%CI: 4.5–8.10 mo)vs6.9 mo (95%CI:5.4–8.8 mo)]. Compared with the development of ICIs and cancer vaccines, adoptive cell transfer therapy is still in the early development phase against pancreatic cancer (Table 3); more preclinical and clinical studies are needed to further explore its full clinical potential.

Table 3 Complete adoptive cell transfer-based clinical trials in pancreatic ductal adenocarcinoma
IMMUNOTHERAPY AND THE TUMOR MICROENVIRONMENT
Immunotherapy has thus far failed to fulfill its promise in PDAC. The underlying mechanisms appear to be complex and multifactorial primarily due to their unique genetic signatures, metabolic features,and immunosuppressive TME. Pancreatic cancers carry unique molecular genetic backgrounds. MSI in pancreatic cancer is extremely rare (approximately 1%). OncogenicKRASmutations, the most common mutation in PDAC, have also contributed to PDAC initiation and maintenance by producing an immunosuppressive TME[129].
Furthermore, altered metabolism of glucose, amino acids, and lipids and their crosstalk with the TME play essential roles in PDAC tumor progression[130]. Multiple lines of evidence have pinpointed the TME as one of the significant barriers to developing effective immunotherapy for PDAC. It is of great clinical interest to sensitize PDAC to immunotherapy through modification of the TME.
One such effort has been focused on CAFs in the TME. As an immunosuppressive component of the TME, FAP-positive CAFs potentially account for the ineffectiveness of immunotherapy in PDAC[131].Another subtype of CAFs, characterized by the expression of the leucine-rich repeat-containing 15(LRRC15) protein, could only be detected in pancreatic cancer tissue and is associated with poor response to anti-PD-L1 therapy[132]. Notably, FAP-positive CAFs are the only CAF subtype that expresses CXC motif chemokine ligand 12 (CXCL12). Ablation of FAP-positive CAFs or inhibition of CXCL12 uncovers the antitumor activity of CTLA-4 and PD-L1-based immunotherapy[133]. A phase I/II clinical trial (NCT03168139) (https://clinicaltrials.gov/ct2/show/NCT03168139) was conducted to evaluate the treatment effect of pembrolizumab in patients receiving docetaxel (NOX-A12), an agent targeting CXCL12 and TME in metastatic PDAC. No results have been reported yet.
Cellular components in the TME, including MDSCs and TAMs, are also promising targets in the combinatory strategy for immunotherapy. MDSCs and TAMs induce an immunosuppressive TME,partially through colony-stimulating factor 1 receptor (CSF1R) and focal adhesion kinase (FAK)[134].Small molecular inhibitors of CSF1R or FAK can reprogram the TME and improve T lymphocytemediated pancreatic cancer destruction[135,136]. Multiple clinical trials with CSF1R or FAK inhibitors combined with immunotherapy are currently ongoing (Table 4).
CONCLUSION
Despite advances in translational research, PDAC remains a highly lethal malignancy. Recent breakthroughs in immunotherapy have revolutionized cancer therapy and have shown great potential to transform future PDAC treatment. However, PDAC has shown inferior treatment outcomes towardvarious immunotherapy regimens compared to other cancer types. The TME has been considered as the fundamental underlying barrier to therapy resistance. To overcome this therapeutic resistance, further investigations with innovative treatment strategies will be needed.

Table 4 Ongoing clinical trials with immunotherapy plus agents targeting the tumor microenvironment in pancreatic ductal adenocarcinoma
FOOTNOTES
Author contributions:Smith C and Yin F collected and analyzed the data, made the tables and figure, wrote and finalized the manuscript; Zheng W, Dong J, Wang Y, Lai J and Liu X critically reviewed the manuscript; and All authors have approved the final manuscript.
Conflict-of-interest statement:The authors declare no competing interests in this study.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:United States
ORCID number:Caitlyn Smith 0000-0001-8944-0480; Wei Zheng 0000-0003-3193-2655; Jixin Dong 0000-0002-2757-4464;Yaohong Wang 0000-0001-5964-0465; Jinping Lai 0000-0001-5365-2481; Xiuli Liu 0000-0001-5791-2017; Feng Yin 0000-0002-8444-1123.
S-Editor:Ma YJ
L-Editor:A
P-Editor:Ma YJ
杂志排行

World Journal of Gastroenterology的其它文章
- Crosstalk between dietary patterns, obesity and nonalcoholic fatty liver disease
- Regulatory T cells and their associated factors in hepatocellular carcinoma development and therapy
- Associations of gut microbiota with dyslipidemia based on sex differences in subjects from Northwestern China
- Prognostic significance of hemoglobin, albumin, lymphocyte,platelet in gastrointestinal stromal tumors: A propensity matched retrospective cohort study
- Contrast-enhanced ultrasound Liver Imaging Reporting and Data System: Lights and shadows in hepatocellular carcinoma and cholangiocellular carcinoma diagnosis
- Isolated gastric variceal bleeding related to non-cirrhotic portal hypertension following oxaliplatin-based chemotherapy: A case report