46XX-17α-羟化酶缺乏症助孕成功1例
2022-08-08张春梅王海宁
张春梅,杨 蕊,李 蓉,乔 杰,王海宁,王 颖△
(北京大学第三医院1.妇产科,生殖医学中心,2. 内分泌科,北京 100191)
先天性肾上腺皮质增生症(congenital adrenal hyperplasia, CAH)是一种由类固醇合成酶缺陷引起的常染色体隐性遗传性疾病,发病率为1/20 000~1/10 000,其中由 17α-羟化酶缺乏(17α-hydroxylase deficiency,17α-OHD)引起的CAH仅占1%[1]。17α-OHD是CYP17基因突变导致17α-羟化酶部分或完全缺乏,17α-OHD使孕烯醇酮不能转化为17α-羟孕烯醇酮和17α-羟孕酮(17α-hydroxyprogesterone,17α-OHP), 从而不能生成足够的皮质醇及性激素,皮质醇不足反馈性地导致促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)分泌过多引起双侧肾上腺皮质增生,性激素不足表现为雌二醇(estradiol,E2)和睾酮(testosterone,T)合成受阻,脱氢表雄酮和雄烯二酮(androstenedione,A)降低,从而引起促卵泡生成素(follicle stimulating hormone,FSH)和黄体生成素(luteinizing hormone,LH)反馈性升高,同时因17α-羟化过程受阻,孕酮(progesterone,P)等底物堆积,引起去氧皮质酮和醛固酮大量增加,促进保钠排钾,出现低血钾、血容量增多、血钠和血压升高,从而抑制了肾素-血管紧张素系统。患者症状轻重可因17α-羟化酶缺乏程度不同而异。
17α-OHD于1966年被首次报道[2],但迄今为止其研究仍较少见。患者社会性别多为女性,按照染色体核型可分为46XX和46XY,其中46XX型更少见。46XY型性腺为睾丸,但因性发育过程中缺乏雄激素,其外生殖器就会自然发育为女性,表现为女性外阴和阴道盲端,但无子宫、输卵管,需手术切除性腺以预防睾丸肿瘤发生[3]。46XX型内外生殖器均为女性,重型临床表现为原发性闭经、无性发育,轻型可有不规律月经来潮,但体内激素水平紊乱,尤其是P升高,存在生育障碍,是需要辅助生殖技术帮助的人群。由于该病发病率低,治疗经验不足,国内外有关该类型患者助孕治疗的报道很少见。现报道1例北京大学第三医院生殖医学中心经过诊治后成功妊娠的病例,以便为此类疾病的诊治提供思路和经验。
1 病例资料
患者女性,34岁,因未避孕未孕7年于2018年9月来北京大学第三医院就诊,患者本人为足月顺产,18岁月经初潮,月经周期规律,5~7 d(28~30 d),孕0产0。患者于2015年曾在外院行体外受精-胚胎移植(invitrofertilization-embryo transfer,IVF-ET)助孕,具体过程和方案不详。自述IVF过程中发现P水平高,最高约16.6 nmol/L,但未能进一步明确病因。最终取卵11枚,因P高在新鲜周期未移植,冻存4枚胚胎。随后给予口服避孕药和促性腺激素释放激素激动剂(gonadotropin releasing hormone agonist,GnRH-a)降调节后,P下降至正常水平,然后冻融胚胎移植(frozen-thawed embryo transfer,FET)2次,均未孕。2016年曾于外院查17α-OHP、皮质醇未见异常,给予地塞米松治疗,服药后P可降至正常,停用地塞米松后P再次升高,之后患者未规律服药和监测。2017年6月曾于外院行腹腔镜下左侧卵巢囊肿剥除术+盆腔粘连松解术,组织病理学诊断提示卵巢子宫内膜异位囊肿。家族史:父母体健,否认近亲结婚史。患者有一姐姐,姐姐已生育2名男孩。查体:身高170 cm,体质量70 kg,血压正常,腋毛及阴毛发育稀疏,乳房发育正常。妇科查体:外阴发育正常,阴毛女性分布,阴道通畅,宫颈光滑,子宫前位、大小未见异常、活动好、无压痛。双附件区未扪及异常。行妇科彩色B超提示子宫大小正常,内膜厚0.7 cm;右侧卵巢3.7 cm×2.0 cm,窦卵泡探及不满意,其内见2个无回声区,大者为2.6 cm×1.2 cm;左侧卵巢3.1 cm×2.1 cm,内见8~9个窦卵泡。实验室检查:2018年9月测FSH 9.52 IU/L,LH 8.89 IU/L,E2 233 pmol/L,T<0.69 nmol/L,A 1.17 nmol/L,P 9.99 nmol/L,泌乳素(prolactin,PRL) 16.9 μg/L,ACTH 升高,为84.9 ng/L(7.2~63.3 ng/L),皮质醇81 μg/L(50~150 μg/L)和17-OHP 0.58 μg/L(0.1~2.3 μg/L)均正常,肾素活性为0.16(0.93~6.56),血管紧张素2(立位)52.88(55.3~115.3),血钾未见异常。染色体核型为46XX,基因检测发现女方CYP17A1基因存在两处杂合突变,分别为C.1459-1467delGACTCTTTC缺失突变和C.1263G>A点突变,均为致病突变(图1),经验证后发现两处突变分别来自其母亲、父亲,为复合杂合突变。同时对男方进行CYP17A1基因筛查未见明确致病改变。
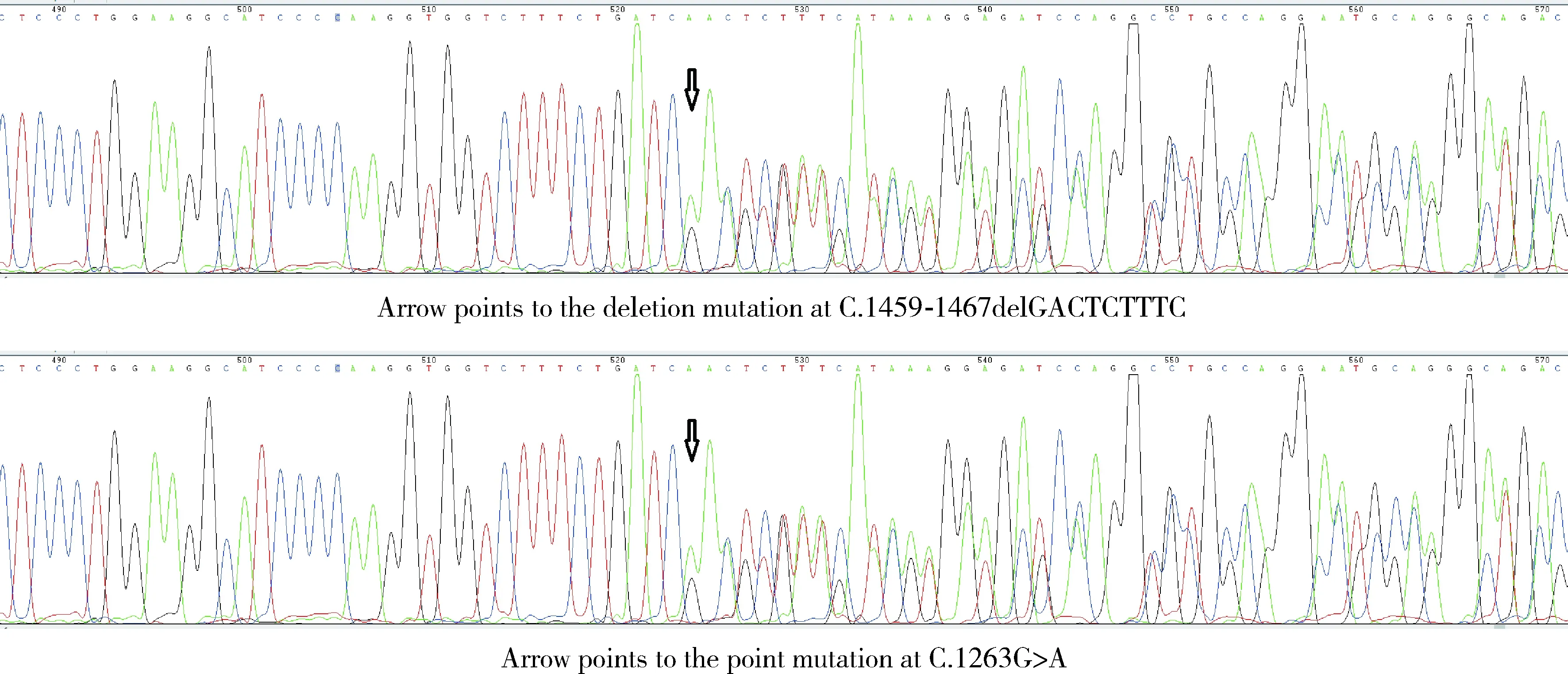
图1 病例CYP17A1基因突变位点Figure 1 CYP17A1 gene mutation sites in the female patient
患者基因检测结果明确,诊断为原发不孕、17α-OHD、IVF-ET失败史。患者内、外生殖器发育正常,仅表现为青春期发育略晚,18岁初潮,但初潮后月经规律,无17α-OHD典型的高血压和低血钾,临床表型轻,评估后认为其有生育机会。患者月经规律,提示可能有自发排卵,P高可能是患者不孕的原因。决定先请内分泌科医师协助控制激素水平,待激素异常纠正后再给予IVF治疗。内分泌科医师给予地塞米松治疗控制ACTH及激素水平,地塞米松用量开始为每日0.375 mg口服,之后增量至早上0.375 mg和晚上0.75 mg,用药1个月后ACTH下降至正常。2019年2月术前准备完成后行IVF-ET,2019年2月14日患者月经第2天行B超检查提示子宫内膜厚0.4 cm,右侧卵巢2.2 cm×1.1 cm,内见2~3个窦卵泡,左侧卵巢3.9 cm×2.8 cm,内见无回声区3.5 cm×2.0 cm,另外见3个窦卵泡,查FSH 7.07 IU/L,LH 8.23 IU/L,E2 141 pmol/L,P 5.15 nmol/L,P偏高,给予炔雌醇环丙孕酮片治疗1个周期,于2019年3月13日复查B超提示子宫内膜厚0.4 cm,右侧卵巢1.9 cm×1.4 cm,3~4个窦卵泡,左侧卵巢4.3 cm×3.3 cm,内见3.1 cm×2.6 cm无回声区以及3个窦卵泡,FSH 11.6 IU/L,LH 6.85 IU/L,E2 227 pmol/L,P 4.29 nmol/L。患者用药后P较前下降,略高于正常,决定给予注射用醋酸亮丙瑞林微球(丽珠制药公司,上海)3.75 mg超长方案降调节。28 d后降调节满意,2019年4月10日查FSH 5.73 IU/L,LH 0.56 IU/L,E2 <73.4 pmol/L,P<0.64 nmol/L,2019年4月13日开始给予控制性促排卵,采用注射用重组人卵泡激素(默克雪兰诺公司,德国)300 IU联合注射用高纯度尿促性素(辉凌公司,瑞士)75 IU方案促排卵13 d,2019年4月27日行取卵术,获卵10枚,常规受精形成6枚胚胎 (胚胎级别8G1×1, 8G2×4,10G2×1)冻存。促排卵后P升高明显,放弃新鲜周期移植(激素水平变化见表1)。2019年5月8日患者月经第3天给予GnRH-a 3.75 mg降调节,降调节后28 d开始人工周期FET,首先给予戊酸雌二醇片4 mg,2次/d,用药7 d,测内膜厚度0.6 cm,加用雌二醇片/雌二醇地屈孕酮片每日1片红片(内含雌二醇2 mg)继续用药7 d,内膜厚度达0.9 cm,随后按北京大学第三医院生殖医学中心常规孕激素转化内膜,于2019年6月21日移植2枚胚胎(8G1×1,5G1×1)。移植术后第13天查血人绒毛膜促性腺激素(human choriogonadotropin,HCG)215.65 IU/L,由于地塞米松在孕期可以通过胎盘影响胎儿,不宜继续使用。考虑病情尚稳定,内分泌科医师建议停用肾上腺皮质激素。移植术后第21天复查血HCG 6 593 IU/L,术后第30天妇科彩色B超检查提示宫内孕,单活胎。人工周期药物于孕12周停药,孕37周因“羊水过少、妊娠期高血压”行剖宫产分娩一男婴,体质量2.94 kg,Apar评分10分(表1)。
2 分析与讨论
17α-OHD非常罕见,编码酶蛋白的基因突变的序列不同会对酶活性造成不同的影响,酶活性的差异将导致不同程度的性发育异常及内分泌系统激素水平的异常[4],因此临床表型程度轻重不同。完全性酶活性缺失的患者症状严重,有的患者甚至不能存活;而部分性17α-OHD酶缺乏到一定程度时出现临床表现,大多数患者表现为闭经,可能会有部分第二性征的发育,多合并高血压和低钾血症。如果酶活性>25%,则患者可能无明显表现而易被漏诊或者误诊[1]。部分性17α-OHD酶缺乏主要就诊原因常为青春期原发性闭经和无乳房发育,其他原因可为查体发现高血压而一般降压药效果差和反复出现卵巢囊肿,青春期出现并且逐渐加重的乏力、软瘫和骨痛等。查体一般体形瘦长、肤色较黑及第二性征不发育。辅助检查血促性腺激素和ACTH升高,E2、雄激素和皮质醇减少,P升高。
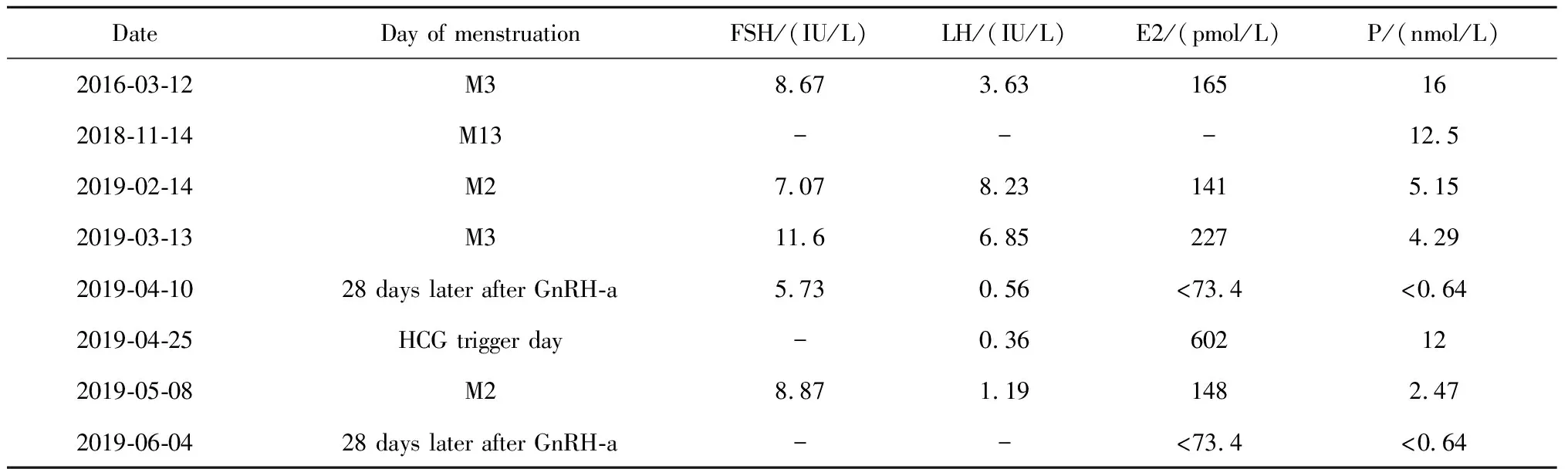
表1 患者的性激素变化Table 1 The changes of gonadal hormones in the patient
FSH, follicle stimulating hormone; LH, luteinizing hormone; E2, estradiol; P, progesterone; GnRH-a, gonadotropin releasing hormone agonist; HCG, human choriogonadotropin.
46XX型 17α-OHD诊断有一定的难度,首先,需要与CAH其他类型相鉴别, 21-羟化酶缺陷症(21-hydroxylase deficiency,21-OHD)是最常见类型,占CAH的90%~95%。21-OHD由于21-羟化酶发生缺陷,将直接导致P和17-OHP转化11-去氧皮质酮及11-去氧皮质醇障碍,进而导致盐皮质激素与糖皮质激素的生成减少,对腺垂体负反馈减少,ACTH水平增加,造成肾上腺增生等一系列临床表现。21-羟化酶的底物(如17-OHP、P、脱氢表雄酮等)增多,通过17α-OHD/17,20碳链裂解酶途径生成过量的雄激素,产生高雄激素相关临床症状,如男性化表现、阴毛过早出现或性早熟、骨龄发育过快等[5]。其次,需要与XY-17α-OHD型患者相区别,二者虽然外生殖器均为女性状态,但46XX型有子宫和卵巢,而 46XY型由于存在性别决定基因导致女性内生殖道形成受阻,子宫、输卵管和阴道上 1/3缺如,通过查染色体核型可以明确诊断。再次,46XX患者的性腺中有原始卵泡,在高促性腺激素的作用下反复出现卵巢黄素化囊肿[6],有时会误诊为卵巢肿瘤而行手术。本例患者多次B超均提示卵巢囊肿,应当考虑卵巢囊肿是受激素影响所致,在肿瘤标记物正常的情况下,给予糖皮质激素治疗的同时可以应用口服避孕药或者GnRH-a治疗,囊肿往往会消失。如果治疗无效或者不能除外恶变可能,必要时谨慎选择手术以明确囊肿性质。此外,患者FSH升高合并卵巢多发囊肿需要与垂体FSH肿瘤相鉴别,后者特征性的激素水平变化为FSH、E2及PRL增高,而LH 降低,早期常无垂体方面的临床表现,而大部分患者卵巢内有多个较大囊肿出现,甚至多次手术均为卵巢滤泡囊肿,直到瘤体增大,出现如视野缺损、头痛等肿块效应后才被发现。多巴胺激动剂有一定的治疗效果,但未发现有药物能够缩小瘤体,目前治疗仍以手术为主[7]。另外,该病需要与卵巢功能减退(decreased ovarian reserve,DOR)相鉴别。DOR患者也表现为FSH和LH水平升高,卵泡数量减少,但P水平正常,并且不会合并高血压或者低血钾等代谢紊乱。
对于核型为46XX的患者,结合生殖器官的发育情况评估是否有生育机会。由于该病非常罕见,迄今为止,国内仅见2例报道,但未介绍助孕过程[8]。国外助孕成功的病例仅见4例:最早一例发表于1995年,为赠卵周期,患者性发育不成熟,只有轻微的第二性征,给予激素替代治疗,经历5次移植后双胎妊娠,然而孕期发生了先兆子痫、HELLP综合征(hemolysis,elevated liver enzymes,and low platelet count syndrome,HELLP syndrome)等严重的并发症[2];2016年巴西学者报道了首例应用自身卵子助孕成功病例,患者为 24岁年轻女性,合并高血压,基因检测提示CYP17A1基因复合杂合突变,采用GnRH-a黄体期长方案IVF,降调节后人工周期FET后成功妊娠[9];第3例由2018年瑞典学者报道,也是目前报道的年龄最大的患者,39岁女性,43岁时行IVF助孕成功[10];2018年日本学者报道了第4例患者,采用长方案,进行了两次人工周期FET,时间间隔了3年,两次移植均成功妊娠分娩[11]。对于该类患者而言,月经规律和乳房发育正常提示酶缺乏程度低,从而临床表型轻。据统计,在17α-OHD患者中有正常乳房发育的妇女仅占13%[12],国外报道的第3例、第4例和本例患者均有规律的月经和正常乳房发育,临床表型轻也是她们妊娠成功最主要的原因。这部分患者也是有可能受益于辅助生殖技术而实现生育愿望的人群。
有关17α-OHD的治疗,首先给予糖皮质激素治疗纠正内分泌紊乱,这是所有治疗的基础,氢化可的松、地塞米松及泼尼松均可作为选择,儿童、青少年治疗首选氢化可的松,青春期后可改为泼尼松或地塞米松治疗。氢化可的松作用时间短, 每天一次通常难以达到良好的抑制作用,需分次服用。地塞米松抑制ACTH的作用最强且持久,因为其影响儿童骨骼发育,不推荐儿童使用。国内报道成年女性患者地塞米松初始剂量为0.75~2.00 mg/d,维持剂量为0.100~0.375 mg/d,能够明显改善症状[13],同时给予人工周期替代以促进第二性征发育。对于有生育机会的患者,在内分泌紊乱得到纠正的情况下可以进行IVF助孕。促排卵过程中需一直应用糖皮质激素控制高P水平,由于地塞米松能够通过胎盘,妊娠后需改为泼尼松治疗,以减少对子代的影响。17α-OHD 患者不孕主要与E2缺乏和高P有关,完全性的E2缺乏卵泡发育阻滞,难以有成熟卵母细胞。从本病例IVF过程中监测的激素水平可见,尽管整个促排卵过程直到扳机日E2水平均较低,扳机日仅为602 pmol/L,但仍有成熟的卵母细胞发育和优质胚胎形成,由此可见,低E2和高P水平并不影响外源性促性腺激素作用下卵母细胞以及胚胎质量。高P主要影响子宫内膜的容受性,因此不能进行新鲜周期胚胎移植,全部冻存胚胎之后选择人工周期或者降调节后人工周期FET即可。本例患者成功受孕后再次请内分泌科会诊,认为患者病情稳定,建议停用肾上腺皮质激素,因此在孕期未继续应用泼尼松治疗。需要注意的是,在进行促排卵之前,需要将性激素控制在理想水平,如激素控制不满意,卵泡可能会出现发育不良从而需要中止周期。在促排卵方案的选择上无特殊,长方案、超长方案或者拮抗剂方案均可。结合国外报道及本病例,长方案或者超长方案可能更容易调整内分泌状态,此外,患者可能存在高FSH,降调节有利于增加FSH受体的敏感性,从而获得较好的妊娠结局。
综上所述,部分性17-OHD 在临床上罕见,极易误诊、漏诊,因此在临床工作中遇到原发闭经、性激素低下,但高孕酮和第二性征不发育的患者,尤其合并高血压、低血钾者,应考虑17α-OHD的可能,并行染色体核型分析及基因测序以进一步明确诊断。由于17α-OHD可能有多系统异常表现,应加强多科室协作及多学科领域相关知识的学习,请相关科室协助排除其他系统疾患及指导治疗。对于有生育要求的部分性46XX-17α-OHD患者,经过评估后如果有生育机会,经过充分的激素调控,借助于辅助生殖技术,患者有希望成功妊娠。
