Genetic Transformation of NGc Insect-resistant Gene in Hi-II Maize
2022-08-05YueLIULidaWANGYingLANQingchaoLIYangLIUXuZHENGXiumeiZHAO
Yue LIU, Lida WANG, Ying LAN, Qingchao LI, Yang LIU, Xu ZHENG, Xiumei ZHAO
Qiqihar Branch, Heilongjiang Academy of Agricultural Sciences, Qiqihar 161006, China
Abstract In order to promote the research of transgenic insect-resistant maize, the target gene were transferred into maize material Hi-II by Agrobacterium-mediated genetic transformation of maize embryos, and maize plants with CryNGc insect-resistant genes were cultured by explant infection, co-culture and differentiation screening to study the genetic expression and resistance of exogenous genes in the offspring. The results showed that the infection effect was the best when the size of young maize embryo was 1.2-1.8 mm. Ten positive transformed plants with CryNGc insect-resistant genes were successfully obtained, and the transformation efficiency was 1.428 ‰.
Key words Maize; Insect-resistant gene CryNGc; Genetic transformation; PCR
1 Introduction
Maize is the second largest food crop in China and is one of the crops with the highest value-added function[1-3]. In order to achieve the strategic goals of food security and ecological security and improving agricultural production efficiency, improving the scientific and technological content of maize varieties is one of the important measures with practical significance[4]. Maize borer is an important insect pest of maize, millet, sorghum and other food crops. Due to the damage of maize borer, the annual loss of maize reaches up to 6 million to 9 million t. The yield of maize is generally reduced by 10%-15% due to the harm of maize borer, and more than 30% or even no harvest in severe years, seriously affecting the yield and quality of maize[5]. In addition to directly harming maize and causing severe losses, the wounds caused by maize borer are also the invasion ways of the pathogens of maize ear rot, and the pest often carries pathogens, which induces and aggravates the occurrence of maize ear rot[6]. Once ear rot occurs in maize, a large number of mycotoxins will be produced in the grain, which will degrade the quality of maize. Studies have found that the incidence of maize ear rot is closely related to the harm of insect pests[7].
The prevention and control technology of maize borer in China has been developed greatly, which has reduced the harm of maize borer to a certain extent. However, as there are many difficulties in the control of maize borer, the actual control area is 20%-30%, and the control effects are different due to different degrees of farmers’ mastery of various technologies[8]. In this study, a novel insect-resistant geneCryNGcwas constructed by domain swapping, including domains I and II ofCry1Aband domain III ofCry1Gc. The plant expression vector was constructed by using novel insect-resistant geneCryNGc, and then transferred into maize material Hi-II byAgrobacterium-mediated method to obtain transgenic positive plants, in order to provide new materials for the study of transgenic insect-resistant maize and to provide a new reference for the development of transgenic insect-resistant maize.
2 Materials and methods
2.1 MaterialsThe plant expression vector pCAMBIA3301 and the test strainAgrobacteriumtumefaciensEHA105 were provided by Heilongjiang Academy of Agricultural Sciences.
The Hi-II maize immature embryos with high regeneration capacity at 9-12 d post pollination were used as transgenic materials. The young embryos with the size of 1.2-1.8 mm were collected on the ultra-clean workbench and disinfected with 75% alcohol for invasion.
2.2 Methods
2.2.1Preparation of culture medium. YEP medium: 5 g/L yeast extract+10 g/L peptone+5 g/L sodium chloride+15 g/L agar, pH 6.8.
Infection medium: 4 g/L N6 basal salt+4 mg/L 2, 4-D+0.7 g/L proline+68.4 g/L sucrose+36 g/L glucose, pH 5.2.
Co-culture medium: 4 g/L N6 basal salt+4 mg/L 2, 4-D+0.7 g/L proline+30 g/L sucrose+8 g/L agar, pH 5.9-6.1; after high-temperature sterilization, the medium was added with 1mg/L AS+1 mg/L N6 vitamin+0.1 mg/L silver nitrate, pH 5.8.
Recovery medium: 4 g/L N6 basal salt+4 mg/L 2, 4-D+0.7 g/L proline+0.5 g/L MES+30 g/L sucrose+8 g/L agar, pH 5.9-6.1; after high-temperature sterilization, the medium was added with 1 mg/L N6 vitamin+1 mg/L Cef, pH 5.8.
Selective medium I: 4 g/L N6 basal salt+4 mg/L 2, 4-D+0.7 g/L proline+0.5 g/L MES+30 g/L sucrose+8 g/L agar, pH 5.9-6.1; after high-temperature sterilization, the medium was added with 1 mg/L N6 vitamin+1 mg/L Cef+0.5 mg/L bialaphos-sodium+0.1mg/L silver nitrate, pH 5.8.
Selective medium II: 4 g/L N6 basal salt+4 mg/L 2, 4-D+0.7 g/L proline+0.5 g/L MES+30 g/L sucrose+8 g/L agar, pH 5.9-6.1; after high-temperature sterilization, the medium was added with 1 mg/L N6 vitamin+1 mg/L Cef+0.5 mg/L bialaphos-sodium+0.1 mg/L silver nitrate, pH 5.8.
GF medium: 4.3 g/L MS basal salt+0.1 mg/L inositol+60 g/L sucrose+3 g/L phytagel, pH 5.9-6.1; after high-temperature sterilization, the medium was added with 1 mg/L MS vitamin+1 mg/L Cef+0.5 mg/L bialaphos-sodium.
Rooting and seedling strengthening medium: 4.3 g/L MS basal salt+0.1 mg/L inositol+60 g/L sucrose+3 g/L phytagel, pH 5.9-6.1; after high-temperature sterilization, the medium was added with 1 mg/L MS vitamin, pH 5.8.
2.2.2Construction of plant expression vector. TheNGcfragment and plant expression vectors were amplified by primers designed based onCryNGcgene, and then digested bySmaI andSacI double enzymes respectively. TheNGcfragment purified from the gel was linked with the fragment of plant vectors, and the connected liquid was transformed into competent cells inEscherichiacoli. After kanamycin resistance screening, the target monoclone was selected for PCR, enzyme digestion and sequencing. The recombinant plasmid was transferred into EHA105 competent cells by alkali lysis.
The plasmid vectors ofA.tumefacienswere extracted by alkali lysis and cultured in the corresponding resistant YEP liquid medium for 24-48 h. Approximately 1.5-5.0 mL of bacterial liquid was centrifuged at 4 ℃, 12 000 r/min for 5 min, and the bacterial cells were collected. The 100 μL of pre-cooling solution I was added, evenly mixed by suction and blowing, and left undisturbed at room temperature for 10 min. Afterwards, 200 μL of solution II was added, reversed 5 times, and cooled on ice for 5 min. The 150 μL of cooling solution III was added, and white floc was produced. The mixture was centrifuged at 4 ℃, 12 000 r/min for 5 min. The supernatant was added with the same volume of phenol/chloroform/isoamylol (25∶24∶1), mixed and centrifuged at 4 ℃, 12 000 r/min for 5 min. The supernatant was then added with the same volume of chloroform: isoamylol (24∶1), mixed and centrifuged at 4 ℃, 12 000 r/min for 5 min. The supernatant was separated, added with twice the volume of anhydrous alcohol (pre-cooling), and evenly mixed. After left undisturbed at -20 ℃ for 30 min, the solution was centrifuged at 4 ℃, 12 000 r/min for 10 min. After the supernatant was discarded, 1 mL of 70% alcohol was added to dissolve the precipitate, which was then centrifuged at 12 000 r/min for 1 min. As the supernatant was discarded, the precipitate was dissolved in 20 μL of sterilized ultra-pure water and then stored at -20 ℃.
PCR was performed in a 2 μL system containing 10 μL of 2×EasyTaqPCRSuperMix, 0.3 μL of each primer, 1 μL of template and 8.4 μL of ddH2O.
The amplification procedures were as follows: pre-denaturating at 95 ℃ for 5 min; denaturating at 95 ℃ for 30 s, annealing at 58 ℃ for 45 s, extension at 72 ℃ for 1 min, 30 cycles; extension at 72 ℃ for 10 min.
Restriction endonuclease reaction of DNA: The recombinant plasmid was digested bySmaI andSacI double enzymes in a 20 μL system containing 8 μL of PCR products, 2 μL of buffer and 1 μL of each restriction endonuclease at 37 ℃ for 1-2 h.
2.2.3Preparation ofA.tumefaciens.A.tumefaciensstored in the -80 ℃ freezer was streaked on YEP solid medium, and single colonies with a size of 1 mm were streaked on YEP solid medium after molecular verification. After cultured at 19 ℃ for 3 d, the bacterial cells were collected and suspended in the infection medium to ensure thatOD550reached 0.3-0.4 for later use.
2.2.4Preparation of explants. The pistil about 9-12 d after pollination was peeled off maize leaves, and soaked in 75% alcohol+20 μL Tween after the top of about 1 cm was cut off. After sterilized on a shaking table at 120-135 r/min for 15 min, the pistil was rinsed twice with sterile water. The young embryos were stripped on the ultra-clean workbench. The young embryos were 1.2-1.8 mm in size and placed in centrifuge tubes containing 2 mL pendant drops. Each tube contained about 100-110 young embryos.
2.2.5Infection of explants. After pendant drops in the centrifuge tubes were discarded, the centrifuge tubes were rinsed with 2 mL of pendant drops, and pendant drops were discarded. The centrifuge tubes were added with 2 mL of pendant drops, slowly inverted and mixed evenly for 20 times, and placed in darkness at room temperature for 5 min. After infection, 1mL ofA.tumefaciensliquid was discarded, and young embryos were placed on sterilized filter paper.
2.2.6Co-culture and recovery culture. The young embryos on the filter paper were inverted at an equal interval on the co-culture medium, so that the convex surface of each young embryo was upward and the flat surface was downward, one tube one dish. The dishes were incubated in darkness at 20 ℃ for 3 d. After 3 d, the complete young embryos, which were not milky white and limp, were transferred to the recovery medium with the convex surface facing up and the flat surface facing down, with about 50 embryos in each dish at an equal interval, and cultured in darkness at 25-28 ℃ for 7 d.
2.2.7Semi-screening culture. After degermination, the young embryos were transferred to selective medium I, the convex surface facing up and the flat surface facing down, with 30-35 embryos in each dish at an equal interval, and cultured in darkness at 28 ℃ for 14 d.
2.2.8Screening culture. All the cultures were isolated and cultured on the screening medium, and the event number was marked. The cultures that did not induce young maize embryos were abandoned, and those induced callus were selected for the next round of screening culture. The resistant tissue larger than soybean grain size was separated from other tissues, and the other tissues were discarded. The resistant callus was screened and cultured in circles for 10-14 d.
2.2.9Dark differentiation. The parenchymal cells with highly differentiated resistance were evenly spaced and loosely distributed in dark differentiation medium, and cultured in darkness at 25 ℃ for 10-21 d until most of the white buds were grown. White buds should be picked out in time.
Light differentiation: White buds were picked into light differentiation medium and exposed to bright light at 25 ℃. When more than half of the regenerated seedlings were unearthed, it entered rooting and seedling strengthening period.
Rooting and seedling strengthening: Strong regenerated seedlings were selected for pruning, and root and callus should be cut completely, but the root and stem should not be injured. The regenerated seedlings were transformed to rooting and seedling strengthening medium, and 3-5 seedlings of the same transformation event were placed in the rooting tank. The seedlings were exposed to bright light at 25 ℃ until 3-4 roots were differentiated before being transferred to the greenhouse.
2.2.10Molecular detection of transgenic plants. Specific primers were designed according toCryNGcsequence, and the annealing temperature was 62 ℃. The specific steps were as follows:NGcgene acquisition → activation and subgeneration ofA.tumefaciens→ sampling of young maize ear with target geneNGc→ disinfection of young ear → preparation of suspension and infection solution → stripping of young embryos → infection and co-culture (A medium) → recovery culture after 3 d (H Medium) → semi-screening culture after 7 d (NS1 medium) → screening culture after 14 d (NS2 medium) →continuous screening culture after 14 d (NS2 medium) →dark differentiation after 14 d (F medium) → light differentiation after 21 d (GF medium) → rooting and seedling strengthening after 5-10 d (R medium) → moving to greenhouse to grow leaves → PCR detection to determine whetherNGcgene was transferred into maize genome.
3 Results and analysis
3.1 Construction of plant expression vectorIn this study, a novel insect-resistant geneCryNGcwas constructed by domain swapping, including domains I and II ofCry1Aband domain III ofCry1Gc(Fig.1A). TheNGcfragment and plant expression vectors were amplified by primers designed based on novel insect-resistant geneCryNGc, digested bySmaI andSacI double enzymes, ligated and transformed to construct recombinant plasmids. As shown in Fig.1B, a band of about 1 000 bp was detected by PCR, which proved that the recombinant plasmid was successfully constructed.
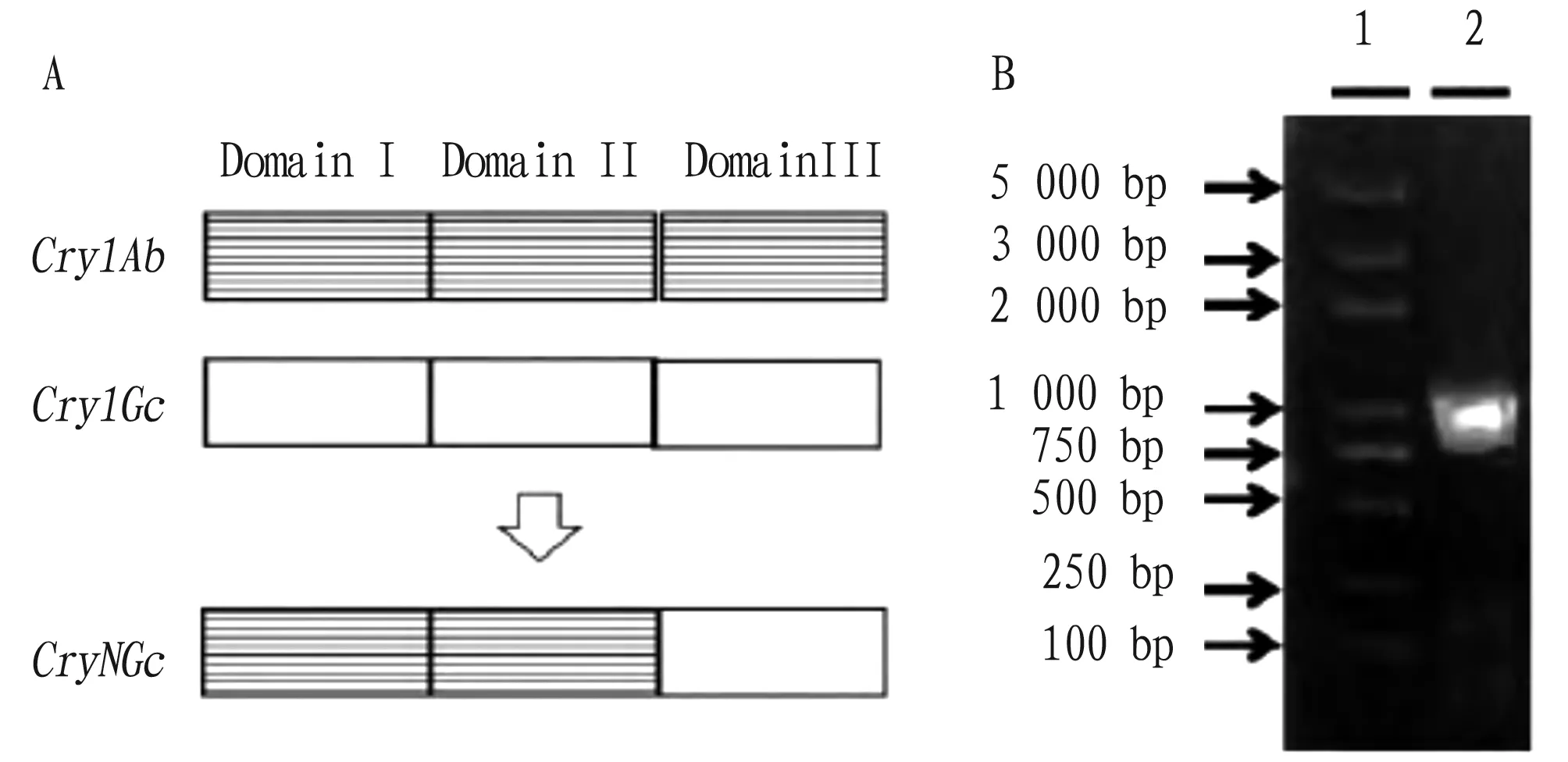
Note: A. CryNGc domain swapping; B. PCR amplification of recombinant vector; 1. 5 000 bp Marker; 2. Plasmid.
3.2 Effect of explant size on genetic transformationThe size of young embryos directly affects the effect of genetic transformation. According to the results of explant infection test, when the size of young embryos was 1.2-1.8 mm, the survival rate of young embryos after infection was the highest of 85%, which was 2.36 times of that of young embryos less than 1.2 mm and 1.54 times of that of young embryos larger than 1.8 mm (Fig.2). Therefore, the size of young embryos was set at 1.2-1.8 mm in subsequent tests. In addition, it was found that the genetic transformation efficiency was higher when young embryos were infected with the bacterial liquid with theOD550of 0.3-0.4.
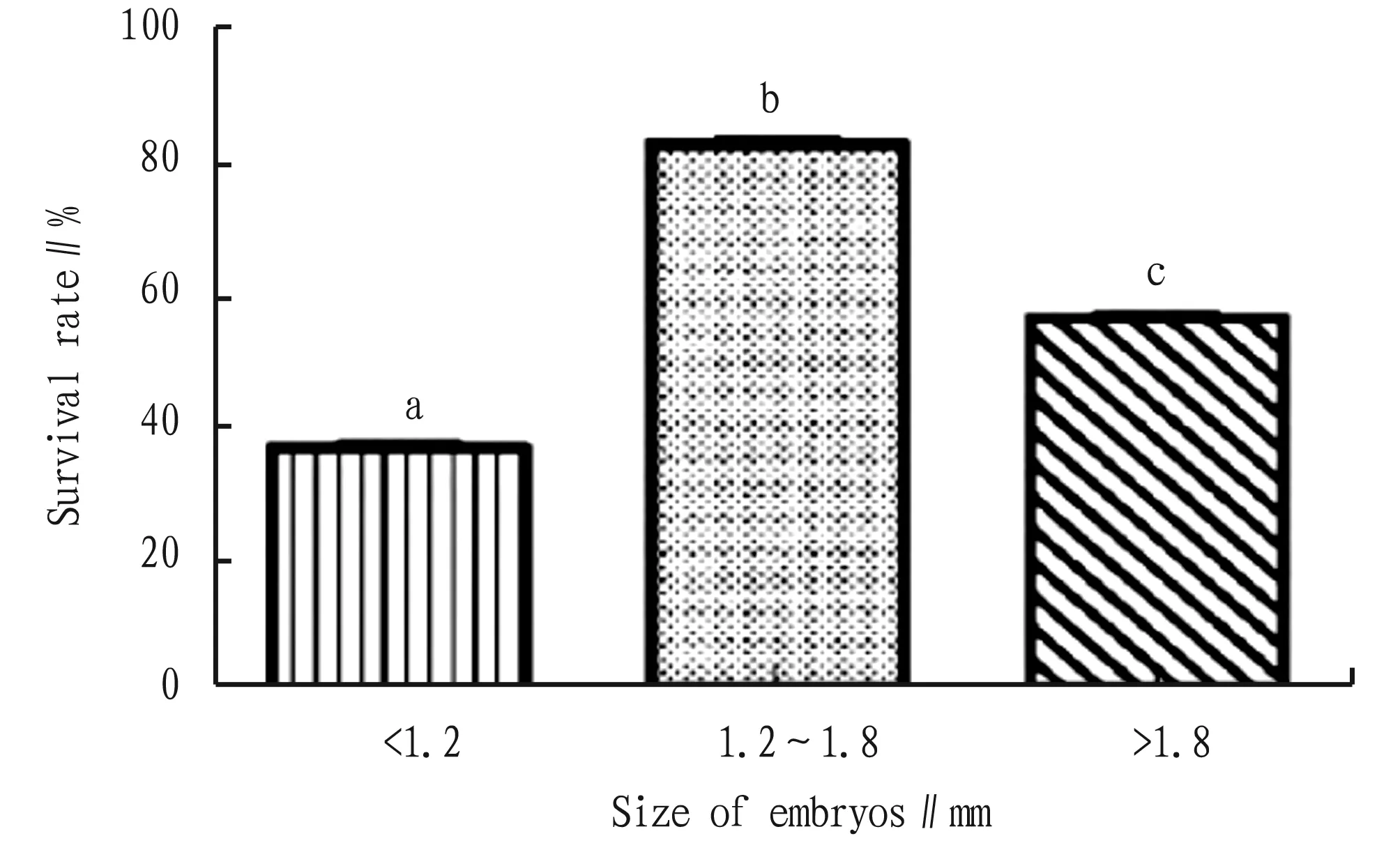
Note: Different lowercase letters represent significant difference at 0.05 level.
3.3 Growth of maize embryos at different stagesAfter young embryos were rinsed with pendant drops, 2 mL of infection solution was added and explants were infected in the dark at room temperature. It was found that the infection effect was the best when the infection time was 5 min. When the infected young embryos were placed upside down on the co-culture medium and the convex surface was upward for co-culture, it was found thatA.tumefacienscould grow only with intact young embryos. Therefore, the integrity of stripped young embryos should be ensured as far as possible during the preparation of explants (Fig.3A and 3B).
In order to promote the growth of young embryos, the young embryos were transferred to recovery medium for recovery culture, and then the recovered embryos were further semi-screened on selective medium I. The results showed that the survival and growth status of young embryos were significantly improved after recovery culture, and the callus was screened at the initial stage after semi-screening culture (Fig.3C and 3D). In order to increase callus screening and eliminate false positive, the callus initially selected was screened on selective medium II, and then the resistant callus was cultured by dark differentiation. It was found that when the diameter of callus reached 2-4 cm, it was better to enter dark differentiation culture, and when the resistant tissue was large and did not enter dark differentiation in this round, it could be decomposed into pieces of 1 cm in diameter for further screening and culture (Fig.3E and 3F).
The resistant callus after dark differentiation culture was transferred to GF medium for light differentiation culture to obtain regenerated seedlings. The regenerated seedlings with strong growth were selected for rooting and seedling strengthening and transferred to greenhouse to grow leaves. In conclusion, resistant callus were obtained by two-round subculture of 0.5 mg/L bialaphos-sodium and one-round decomposition of 1.0 mg/L bialaphos-sodium, and transformed seedlings were obtained by light culture.
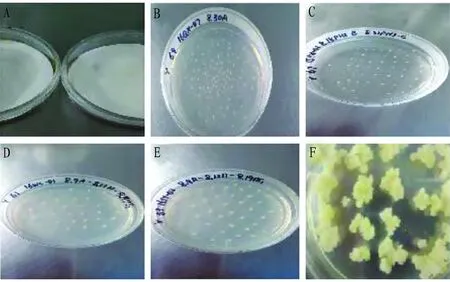
Note: Contact the concave surface of the embryo with A medium, with the convex surface facing upwards; B. Co-culture; C. Recovery culture; D. Semi-screening culture; E. Screening culture; F. Dark differentiation.
3.4 Detection of transgenic plants by PCRWhen the transformed seedlings were transplanted and survived, DNA was extracted from the leaves for PCR detection. The results showed that a total of 7 000 young embryos were stripped, and the band size of the known target genome was 1 017 bp. The same target fragment was amplified from 10 regenerated seedlings as that of the positive control (Fig.4), indicating that the exogenous geneCryNGchad been successfully transformed into the maize genome, and the transformation efficiency was 1.428 ‰.

Note: 1. 2 000 bp Marker; 2. Positive control; 3. Negative control; 4. Transgenic regenerated seedlings.
4 Discussion
Novel genes have the insecticidal nature of wildBtgene. For example,Cry1andCry2genes have insecticidal activity against Lepidoptera pests;Cry3,Cry7andCry9genes have insecticidal nature against Coleoptera pests;Cyt1,Cry10andCry11genes have insecticidal nature against Diptera pests;Cry5,Cry12andCry14genes can kill nematodes. However, wild-type strains have some disadvantages, so it is expected to obtain new genes by means of molecular modification. Studies have shown that a novel gene namedCry1A.105has been transformed into the transgenic maize variety MON89034, which recombines domain II ofCry1AbandCry1Acand domain III ofCry1F[9-11]. In this study, domains I and II ofCry1Aband domain III ofCry1Gcwas used. In order to verify whether the gene has insect resistance, PCR detection showed thatCryNGcgene was successfully transformed into the maize genome. Inoculation tests were carried out in indoor and outdoor, and it was concluded thatCryNGcgene had strong resistance to Asian maize borer.
In the analysis ofBtgene resistance, polymerase chain reaction (PCR) is the most commonly used method to check whether exogenous target genes are integrated into transgenic plants, which is easy to operate and can qualitatively detect[12]. Therefore, PCR is used to detect the introduction of target genes.Agrobacterium-mediated method is generally adopted for maize genetic transformation. Such transformation system uses natural transformation vector system, which has high incidence of transformation and good transformation results.Agrobacterium-mediated transformation has the advantages of simple transformation and selection, and has been widely used due to its high transformation efficiency and convenience[13]. In this study, a total of 7 000 young embryos were stripped and 10 positive transgenic plants withCryNGcresistance genes were obtained, which may be related to the reduction of survival rate due to mechanical damage of young embryos in the process of stripping or infection. Subsequent experiments will further optimize the genetic transformation conditions to improve the transformation efficiency. At present, the transgenic insect-resistant maize widely used in the world is mainly expressing crystal protein, and these proteins can specifically kill egg white of pests. Studies of the effect of transgenic insect-resistant crops on some aboveground arthropods points out thatBtgene expressed in transgenic insect-resistant crops has specificity of insecticidal spectrum and has no harm and influence on natural enemies of pests, and due to the planting of transgenic insect-resistant crops, the dose of insecticide has been significantly reduced, which also provides a scientific basis for the large-scale promotion of organic green farmland[14-16]. The analysis of insect resistance ofCryNGcgene lays a foundation for the establishment of insect-resistant transgenic maize. In the follow-up experiments, the insect resistance test ofCryNGctransgenic maize will be carried out in the field, in order to obtain more abundantBtgene resources and to further promote the industrialization research of insect-resistant transgenic maize.
5 Conclusions
In this study, a novel insect-resistant geneCryNGcwas constructed by domain swapping method, and then the target gene was transferred into maize material Hi-II byAgrobacterium-mediated genetic transformation of young maize embryos. A total of 7 000 young embryos were extracted, and transformed seedlings were positive in PCR detection. Ten transgenic plants withCryNGcinsect-resistant gene were obtained, and the transformation efficiency was 1.428 ‰, which indicated thatCryNGcresistance gene had been successfully integrated into maize genome, thus laying a foundation for the study of transgenic insect-resistant maize.
杂志排行

植物病虫害研究(英文版)的其它文章
- Occurrence Regularity and Life History of Cigarette Beetle Lasioderrma serricorne (Fabricius) in Tobacco Leaf Threshing and Redrying Workshop
- Observation on Behavior of Chilades pandava
- Field Identification and Prevention and Control Techniques of Citrus Huanglongbing of Orah Mandarin
- Field Control Effects of Four Biopesticides on Grape Gray Mold
- Complete Genome Sequence Analysis of Guangdong Isolates of Cymbidium Mosaic Virus
- Effects of EDTA and Zn Stress on Physiological Characteristics of Medicago sativa L.