脊髓小脑共济失调3型临床特征及遗传学分析
2022-08-03潘佳丽刘举魏芷桦杨晓艳陈晨刘洪波
潘佳丽,刘举,魏芷桦,杨晓艳,陈晨,刘洪波
(郑州大学第一附属医院 a.神经内科;b.遗传与产前诊断中心,河南 郑州 450052)
脊髓小脑共济失调(spinocerebellar ataxia,SCA)是以小脑和脊髓某些部位进行性变性为特征的遗传性神经系统疾病,流行病学研究表明,SCA的患病率为每10万人0~5.6人,平均每10万人2.7人,根据基因、染色体位点可分为多种亚型,目前已被描述的有40多种,其中SCA3型是全球最常见的显性共济失调[1]。脊髓小脑共济失调3型(spinocerebellar ataxia type 3,SCA3)又叫做马查多-约瑟夫病(Machado-Joseph disease,MJD),其主要病理损害表现为脊髓和小脑进行性退行性改变,同时伴有脑干、黑质和基底节变性[2],多中青年或成年起病,既往报道,SCA3的发病年龄(age at onset,AO)为4~70岁,但多发生在30~40岁,病程进展缓慢[3]。SCA3主要的临床表现是进行性小脑性共济失调,还可伴有其他非典型临床症状如锥体外系表现、构音障碍、眼球震颤、舌肌束颤和萎缩、锥体束征、周围神经病变、焦虑抑郁等。近年来,关于河南地区SCA3的报道并不多见。本研究收集了36例 SCA3患者的临床资料,分析其临床特征,并对其中一典型家系进行临床表型及遗传学分析。
1 资料与方法
1.1 一般资料收集2016年1月至2021年9月在郑州大学第一附属医院确诊为SCA3的36例患者。纳入标准:符合Harding[4]标准,并由遗传性共济失调基因检测确诊。该家系中,有类似症状但未行基因检测者,参考Harding标准进行临床诊断。排除标准:未进行基因检测的疑似患者,资料不完整;其他原因引起的小脑共济失调,如脑血管病、炎症、小脑肿瘤、中毒等。
1.2 基因检测应用荧光引物标记聚合酶链反应(polymerase chain reaction,PCR)及毛细管电泳技术对所有患者进行脊髓小脑共济失调常见亚型SCA1、SCA2、SCA3、SCA6、SCA7、SCA8、SCA12和DRPLA致病基因检测,且对本研究一家系成员进行SCA3基因CAG动态突变检测。采集患者外周静脉血4 mL,用EDTA抗凝,采用全血基因组DNA抽取试剂盒(美国OMEGA公司)提取基因组DNA作为PCR扩增模板。引物设计、PCR扩增以及DNA测序由上海生工生物工程公司完成。
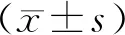
2 结果
2.1 SCA3患者临床特征男8例,女28例,男女比为2∶7,均为杂合子。发病年龄20~63(39.72±11.00)岁,病程1~20 a,平均8(4,9)a。SCA3基因CAG重复55~79(64.86±5.09)次,发病年龄与CAG重复次数呈负相关(r=-0.762,P<0.001),病程与CAG重复次数呈正相关(r=0.331,P=0.049)。
30例(83.3%)以步态不稳伴头晕为首发症状,仅有3例(8.3%)以言语不清、饮水呛咳为首发症状,1例以双下肢麻木为首发症状,1例以头部震颤为首发症状,1例以双下肢无力伴疼痛为首发症状。所有发病者均有步态不稳和小脑共济失调症状,言语障碍、饮水呛咳、吞咽困难可与走路不稳同时出现,也可在发病数年后逐渐出现,病情均呈进行性加重。25例(69.4%)患者有家族史,21例(58.3%)患者病理征阳性,22例(61.1%)患者腱反射活跃或亢进。头颅磁共振结果显示有脑萎缩13例(36.1%),最常见于小脑半球,其次为脑干,多数为轻度小脑萎缩。见图1、2、3。

图1 36例SCA3患者临床表现
图2 SCA3患者的神经系统检查
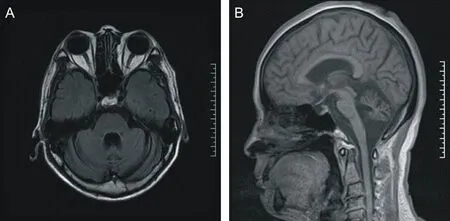
2.2 典型家系临床资料先证者,男,36岁,以双下肢无力伴震颤为首发症状就诊于郑州大学第一附属医院神经内科,诉病史3 a,起初表现为打篮球时跑跳障碍,双下肢不自主震颤,抬脚吃力。6个月前症状逐渐加重,表现为走路不稳,左右摇晃如醉酒样。神经系统查体:双眼无眼球震颤,四肢肌肉无萎缩,双下肢肌力4级,双上肢肌力5级,双下肢肌张力稍增高,双上肢肌张力正常,双侧膝反射活跃,Romberg征阳性,双侧跟-膝-胫试验欠稳准,深浅感觉未见异常,病理征未引出,呈宽基底步态,走“一字”不稳。头颅MRI提示小脑脑沟稍宽,轻度小脑萎缩待排除;磁敏感加权成像未见明显异常。实验室检查未见明显异常。追问病史,描述其外祖母、母亲、舅舅、姨母等共13人有走路不稳的家族史,对该家系成员进行了调查,见表1,家系图见图4。家系中6位成员完成了SCA3基因筛查,结果见表2。5代人共14人发病,其中2例确诊,12例临床诊断,3例症状前患者。SCA3基因检测毛细管电泳结果见图5。
A为颅脑MRI T2加权像,示双小脑半球萎缩;B为颅脑矢状位MRI T1加权像,示小脑、脑桥萎缩。

表1 家系中患病成员临床特征
表2 家系成员SCA3基因重复次数
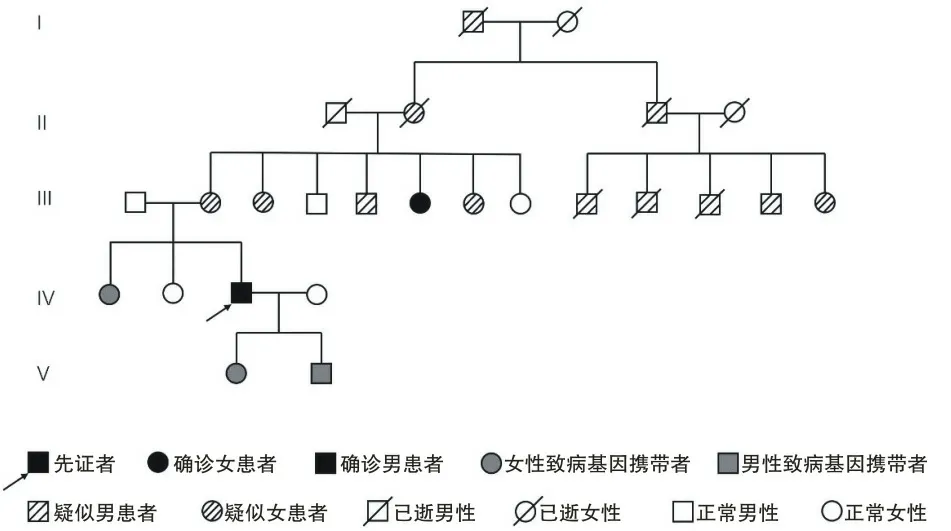
图4 SCA3家系图

A.先证者(Ⅳ3)基因检测结果(毛细管电泳方法),CAG重复数目为20次和62次;B.先证者三姨(Ⅲ5)重复数目为7次和61次;C.先证者大姐(Ⅳ1)重复数目为20次和63次;D.先证者二姐(Ⅳ2)重复次数为5次和8次;E.先证者女儿(Ⅴ1)重复数目为13次和62次;F.儿子(Ⅴ2)重复数目为13次和63次。箭头示异常CAG重复片段。
3 讨论
SCA3是我国最常见的脊髓小脑共济失调亚型,其致病基因是位于14号染色体长臂(14q24.3 -32)上的ATXN3基因。SCA3的发病是由于ATXN3基因的第10外显子区域的三核苷酸CAG重复扩增,神经元多聚谷氨酰胺ATXN3蛋白聚集引起蛋白构象改变而导致的。因此,在遗传学上SCA3属于动态重复扩增突变,也称为多聚谷氨酰胺疾病[2]。ATXN3基因编码ataxin-3蛋白,ataxin-3是一种去泛素酶,参与多种细胞过程,如去泛素化、细胞骨架组织和转录调控。正常的ataxin-3主要存在于神经细胞的细胞质中,突变的ataxin-3聚集在神经细胞核内,并在大脑许多区域的神经细胞内聚集形成核内包涵体[5]。多聚谷氨酰胺疾病常见的病理特征是疾病蛋白在神经元内的积累和聚集,最常发生在神经元的核内,直接或间接产生神经毒性效应,潜在地损害各种细胞过程和细胞内稳态,最终导致神经元的丢失和脑萎缩[6]。正常ATXN3基因CAG重复次数为12~49次,当CAG重复次数达到51次时可导致SCA3,目前研究表明CAG异常重复大多在51~86次[7]。
SCA3的一些临床、影像学和病理研究已经证实了广泛的神经退行性变,包括大脑皮质、基底神经节、丘脑、小脑和脑干,且脑结构和功能的损伤早于临床表现。SCA3型脑萎缩相对较轻,本研究中仅有少数(36.1%)患者MRI发现了小脑或脑干的萎缩,多为小脑半球萎缩,病情严重者可出现脑桥和延髓的萎缩。多数(69.4%)SCA3患者具有家族史,少部分为散发型。此外,CAG重复次数与发病年龄呈负相关,与病程呈正相关,与既往研究结论一致,即CAG重复次数越多,发病年龄越早,疾病越严重[8]。Leotti等[9]的一项纵向研究发现,CAG重复扩增次数与更快速的国际合作共济失调量表评分进展呈正相关,重复序列在70~75次的患者疾病进展更快,并发现30%的进展变化可以用CAG重复长度来解释。该家系的发病成员中,8男6女,发病年龄33~60岁,病程1~20 a,Ⅰ~Ⅲ代在发病6~20 a后去世。先证者以双下肢无力、震颤起病,伴有双下肢肌张力障碍,逐渐出现步态不稳,共济失调,家系中其他成员以行走困难起病,进行性加重,部分出现肌张力障碍但无双下肢震颤。锥体外系表现在SCA3患者中并不常见,容易误诊为帕金森综合征。有研究发现18.1%的SCA患者合并震颤,并且有震颤的SCA患者比无震颤的患者有更严重的共济失调[10]。该先证者年轻起病,并且有共济失调家族史,结合影像学、神经系统查体,应考虑诊断SCA3,经基因检测确诊为SCA3。因此,以肢体震颤为首发症状的SCA3患者要注意与帕金森综合征相鉴别。
本研究发现该家系成员的异常CAG重复扩增次数接近,均在61~63次,但疾病的严重程度不同且存在一定的遗传早现现象,即家系中发病年龄逐代提前,第1代发病年龄在60岁左右,至第4代先证者提前到33岁。这可能与文献报道的结论一致:CAG重复次数并不能完全解释所有SCA3患者发病年龄和严重程度的差异,AO中只有平均55.2%的变异可归因于CAG的扩展程度,提示了其他因素如环境、基因多态性等也在起作用[11]。Mergener等[12]研究发现rs3512的C等位基因对SCA3有保护作用,在相同CAG扩增长度条件下,携带rs3512 G/G基因型的受试者比携带至少1个C等位基因的受试者的发病时间平均早2.44 a。Jiao等[13]研究发现年龄是SCA3疾病严重程度的独立调节因素,在控制其他因素相同的条件下,年龄越大,疾病越严重。
值得注意的是,先证者的大姐,43岁,SCA3基因CAG异常重复数为63次,与先证者(62次)接近,但临床并未发病,考虑可能原因:(1)环境、性别等因素造成的差异;(2)表观遗传的变化可能也参与了发病年龄的发病机制。先前研究显示,同一家系中CAG重复数相同但发病年龄较早的成员ATXN3启动子区域第1个CpG岛的DNA甲基化水平较高,推测可能是ATXN3基因启动子区域高甲基化水平所触发的某些特定通路的激活或抑制[14]。先证者儿子、女儿均被基因诊断为SCA3,但目前均未临床发病,考虑可能是由于该病一般成年发病,发病年龄在30~40岁,先证者女儿14岁,儿子9岁,所以可能还未到发病年龄,以后还需要长期随访观察。本研究还发现,在该家系的5代成员中,父系遗传的后代均临床发病或未发病但基因诊断为SCA3型,母系遗传的后代中部分为正常个体,在既往的家系报道中未观察到此现象,是否可以用既往研究结论来解释:对SCA3基因动态突变的分析发现,亲本对CAG重复传播有显著影响,这种不稳定性在父系遗传中更显著[15]。Pearson等[16]认为可能是精子在有丝分裂周期中减数分裂前突变的结果。
治疗方面,基因修饰治疗是最佳的方法。目前开发的一些新的方法包括在DNA或RNA水平上靶向ATXN3基因,如基因组编辑、新发现的可以与扩展的CAG重复结合的小分子[17],激活蛋白质量控制途径以及细胞稳态等。这些方法的目的在于沉默ATXN3的表达,减少ATXN3的剪切和聚集,激活蛋白质量控制途径和增加细胞稳态[18]。最近的研究发现,反义寡核苷酸靶向RNA的方法,可以有效地降低SCA3小鼠大脑中ATXN3突变体水平[19-20],该方法已处于开发晚期,有望成为新的疾病修饰疗法。但这些方法仍处于临床前阶段,因此该病目前尚无法治愈,临床上无特效药,但对症支持治疗如物理治疗、康复训练、刺激治疗、心理治疗等对改善患者症状、提高患者生活质量、延缓残疾进展、延长患者预期寿命有重要作用。关于纯合子SCA3的报道显示,纯合子SCA3的AO早于杂合子SCA3,且ATXN3纯合子的基因剂量效应可增强临床表型严重程度[21]。因此要避免近亲结婚,通过遗传咨询和产前诊断来阻止突变体ATXN3向下一代的传播。
SCA3是临床异质性显著的遗传性疾病,典型表现是进行性小脑性共济失调,对于非典型症状起病的患者,如肢体震颤,临床医生应详细询问家族史并进行基因突变筛查,以提高疾病诊断的准确率。
