碱性蛋白酶控制酶解诱导大豆分离蛋白纳米颗粒的形成机制
2022-08-02常方圆赵谋明周非白
钟 敏,常方圆,赵谋明,周非白*
(华南理工大学食品科学与工程学院,广东 广州 510640)
生物酶解是一种绿色有效的蛋白质改性手段,可以显著提高蛋白质在抗氧化、调节血脂水平与免疫力等方面的生理活性,通过控制酶解程度还可以提高蛋白质的功能特性,如溶解性、发泡性、乳化性与凝胶性等,是蛋白质精深加工领域的研究重点。近年来研究发现,蛋白质在酶解过程中形成的两亲性结构可自组装形成多种纳微结构,如纤维、纳米管、胶束、球形颗粒等,在食源性功能因子的稳态及新型蛋白基食品体系的构建等方面显示出极大的应用潜力,拓宽了蛋白质在功能性食品领域的开发及应用。
酶解诱导蛋白自组装受到多种因素的共同影响,如底物蛋白的特性、酶的特异性、水解程度、外界环境等。研究发现pH值、蛋白质浓度和Ca都会影响部分水解的-乳清蛋白组装形成纳米管,分别影响纳米管的形成速度、数量及结构的硬度;Gao Yuzhe等发现分别经胰蛋白酶、蛋白酶A、胃蛋白酶和蛋白酶M处理的乳清蛋白形成纳米纤维结构的能力不同;Nicklas等利用碱性蛋白酶对胶原蛋白处理18~168 h,发现控制酶解时间可以得到不同粒径的胶原蛋白纳米颗粒。目前利用生物酶解技术诱导蛋白组装的研究多集中于动物蛋白。EATLancet委员会于2019年发布首份全球健康膳食和可持续食物生产的通用科学标准,对植物蛋白质的开发与应用提出新要求,其中大豆蛋白作为优质植物蛋白来源受到广泛关注。与乳蛋白等动物蛋白相比,天然大豆蛋白组分复杂,球状结构致密,疏水区域分布较广,而且不同亚基酶解敏感性不同,使得各组分酶解过程不同步且酶解程度不易控制。如大豆球蛋白(11S)中碱性亚基大多被包埋在酸性亚基内部,亲水外侧酸性亚基通常优先被降解,而相关研究却发现11S酶解诱导聚集体的形成主要来源于碱性多肽链的肽段;Inouye等对比研究了-伴大豆球蛋白(7S)和11S的酶解聚集规律,发现在中性条件下,11S酶解产生的肽段比7S的酶解肽段更容易聚集,而Kuipers等发现7S具有抑制11S酶解肽的聚集效果。酶解过程中亚基的暴露与降解程度、所生成肽段的序列与带电性以及各组成成分之间的相互作用均影响蛋白的组装行为,如何利用生物酶解技术构建有序的大豆蛋白基纳微结构仍待进一步研究。
课题组前期研究结果表明,大豆蛋白在酶解过程中产生的多肽类结构经超声诱导可进一步组装形成约100 nm的大豆肽基纳米颗粒;进一步研究发现,利用风味蛋白酶、碱性蛋白酶以及复合蛋白酶控制酶解大豆蛋白结合热作用可以成功制备蛋白纳米颗粒,其中酶种类和水解度(degree of hydrolysis,DH)对颗粒的形成都极为关键,而碱性蛋白酶作用下大豆蛋白的组装行为具有特异性。碱性蛋白酶作为目前应用最广的一种商业酶制剂,常应用于改善与提升蛋白质的营养和功能性,如碱性蛋白酶酶解大豆蛋白可以显著提升其抗氧化活性,并且有效减轻炎症症状。此外,碱性蛋白酶控制酶解蛋白可形成更多疏水性单元结构与更小粒径的两亲性肽段,在构建多功能纳微尺度的生物材料方面具有较大潜力,其作用下蛋白的组装行为及机制值得探究。因此,在前期研究的基础上,以碱性蛋白酶作为大豆分离蛋白(soy protein isolate,SPI)原料的水解酶,探究SPI的结构变化规律,进一步明晰其自组装形成大豆蛋白纳米颗粒(soy protein nanoparticles,SPNs)的机制,以期为新型大豆蛋白基功能载体的构建提供一定理论指导。
1 材料与方法
1.1 材料与试剂
低温脱脂豆粕(蛋白质约45%) 山东禹王实业有限公司;碱性蛋白酶2.4 L(酶活力>200h10U/g) 丹麦诺维信公司;十二烷基硫酸钠(sodium dodecyl sulfate,SDS)、二硫苏糖醇(dithiothreitol,DTT)、尿素 美国Geniview公司;邻苯二甲醛(-phthalaldehyde,OPA)、8-苯胺-1-萘磺酸(8-anilino-1-naphthalenesulfonic acid,ANS)、2,2’-联氮双(3-乙基苯并噻唑啉-6-磺酸)(2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid),ABTS)、荧光素二钠、Trolox 美国Sigma-Aldrich公司;蛋白质预染Marker(10~180 kDa) 广州鼎国生物技术有限公司;其他化学试剂均为国产分析纯。
1.2 仪器与设备
THZ-82A恒温振荡器 常州澳华仪器有限公司;pHS-25 pH计 瑞士Mettler Toledo公司;CR22N高速冷冻离心机、HT7700场发射透射电镜、F7000荧光分光光度计 日本Hitachi公司;Alpha 2-4 LDplus真空冷冻干燥机 德国Martin Christ公司;Nano-ZS纳米粒度及Zeta电势分析仪 英国Malvern公司;Chirascan圆二色光谱(circular dichroism,CD)仪 英国Applied Photophysics公司;Mini-PROTEIN 3 Cell电泳仪 美国Bio-Rad Laboratories公司;C300化学发光分子成像系统美国Azure Biosysterms公司;UV754N紫外-可见分光光度计 上海佑科仪器仪表有限公司。
1.3 方法
1.3.1 SPI的提取
采用常规碱溶酸沉的方法:将低温脱脂豆粕粉碎成细小的粉末,过筛收集。豆粕粉与去离子水按1∶10的质量比进行分散,室温搅拌2 h并且过程中维持pH 8.0左右。然后10 000h、4 ℃离心20 min,过滤收集上清液,调节pH 4.5,4 ℃静置30 min后,按照上述条件进行离心,弃去上清液,并用去离子水清洗沉淀表面2 次,加入沉淀质量7 倍的去离子水缓慢搅拌,维持pH 7.5左右,蛋白充分分散后于4 ℃透析48 h,冷冻干燥备用。
1.3.2 碱性蛋白酶酶解处理SPI
准确称取一定量的SPI于去离子水中配成40 mg/mL的蛋白分散液,室温搅拌分散2 h充分水化,加入碱性蛋白酶酶解。酶解条件:pH 8.0,酶解温度55 ℃,加酶量为蛋白质量的0.1%。于固定时间(10 min、30 min、1 h、2 h、4 h、8 h、12 h、24 h)取样,样品调节pH 7.0后立即置于沸水浴(100 ℃)灭酶10 min。样品冷却至室温后10 000h、20 ℃离心15 min,收集上清液,直接用于测试或冷冻干燥备用。
1.3.3 DH的测定
参考Nielsen等的方法,具体步骤为:取400 μL样品(0.2 mg/mL)与3 mL OPA试剂混合均匀,避光准确反应2 min后于340 nm波长处比色,读数记为,标准品和空白对照分别用0.951 6 mmol/L丝氨酸溶液和纯水代替,测定结果记为和。样品中的游离氨基表示为丝氨酸氨基当量(Ser NH),DH计算公式如下:

式中:为样品蛋白质量浓度/(mg/mL);和分别为0.970和0.342 mequv/g;为蛋白质的总肽键数,约为7.8 mequv/g。
1.3.4 粒径、多相分散系数及Zeta电位测定
采用动态光散射(dynamic light scattering,DLS)技术与电泳光散射技术,将样品溶液用10 mmol/L,pH 7.0的磷酸盐缓冲液(phosphate buffered saline,PBS)稀释至0.5 mg/mL后,用Nano-ZS纳米粒度仪对样品的粒径、多相分散系数(polydispersity index,PDI)及Zeta电位进行测定。所有测定均在25 ℃条件下进行,每个样品至少测定3 次。
1.3.5 场发射扫描电镜(field emission scanning electron microscope,FE-SEM)分析
将样品冻干粉末均匀薄涂一层固定于导电双面胶上,喷金后通过加速电压为5 kV的FE-SEM进行微观形貌观察。
1.3.6 十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDSPAGE)分析
参考Laemmli的方法:将样品分别与还原性样品缓冲液(2 g/100 mL SDS,25%甘油,14.4 mmol/L-巯基乙醇,0.05%溴酚蓝;溶于60 mmol/L pH 6.8 Tris-HCl缓冲液)以及非还原样品缓冲液(不含-巯基乙醇,其他同还原性样品缓冲液)混合,控制蛋白质最终进样质量为20 μg。凝胶电泳在恒流25 mA下进行,分离胶和浓缩胶的丙烯酰胺体积分数分别为12%和5%。电泳结束后,进行染色与脱色处理,最终在凝胶成像系统上进行成像拍照。
1.3.7 表面疏水性测定
参考Haskard等的方法,使用ANS作为荧光探针进行测定。具体操作步骤为:将蛋白样品分散于PBS溶液(10 mmol/L,pH 7.0)中并在室温下搅拌1 h,然后梯度稀释配制0.01~0.5 mg/mL的蛋白分散液。向4 mL样品中加入20 μL ANS溶液(8 mmol/L)并混合均匀,立即通过荧光分光光度计测定样品的荧光强度,记为FI;未加入ANS样品的荧光强度,记为FI。设定激发波长为370 nm,发射波长为465 nm,电压为700 mV,激发和发射的狭缝均设置为5 nm。以FI和FI的差值作为轴,蛋白质浓度作为轴,通过线性回归分析计算出的斜率即为表面疏水性。
1.3.8 临界聚集浓度(critical aggregation concentration,CAC)的测定
采用芘荧光探针光谱法测定。准确称取一定量的芘,用适量乙醇溶解后转入容量瓶中,定容并摇匀,制得浓度为1h10mol/L的芘乙醇贮备液。取200 μL芘乙醇贮备液于玻璃试管中,于25 ℃烘箱中挥发干乙醇,加入2 mL不同浓度的样品溶液,超声10 min,静置30 min,测定各溶液中芘的荧光发射光谱。激发波长为335 nm,发射波长为350~500 nm,激发和发射狭缝宽分别5 nm和2.5 nm。
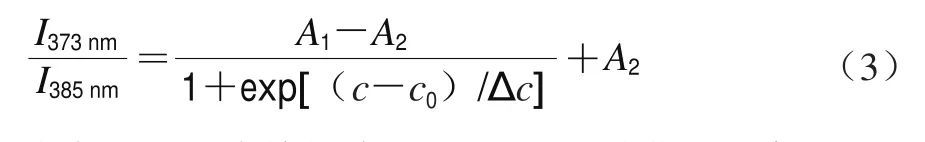
式中:为特征峰1(373 nm)的荧光强度;为特征峰3(385 nm)的荧光强度;为低蛋白质量浓度下的/;为高蛋白质量浓度下的/;为样品浓度;为曲线突变的中点,即蛋白的CAC;Δ为描述Boltzmann曲线突变程度的一个参数,即在突变中点处作曲线的斜率,与、两条平行于轴的直线相交,两交点水平距离的1/4值即为Δ。
1.3.9 二级结构分析
1.3.10 相互作用力分析
参考Liu Fu等的方法,将1 g/100 mL蛋白样品分散于不同变性溶剂(30 mmol/L DTT、0.5% SDS和6 mol/L尿素)或其组合中,作用30 min,观察蛋白样品分散液的外观并测定样品粒径的变化,粒径测定方法参考1.3.4节。
1.3.11 ABTS阳离子自由基清除能力测定
参考Zheng Lin等的方法,将7 mmol/L ABTS溶液与2.45 mmol/L过硫酸钾溶液混合,室温避光反应12~16 h制得ABTS阳离子自由基储备液。将ABTS阳离子自由基储备液稀释一定倍数配成工作液。将50 μL蛋白样品分散液(0.2 mg/mL左右)与150 μL ABTS阳离子自由基工作液混合,30 ℃反应30 min后,使用全波长扫描多功能读数仪于734 nm波长处测定吸光度。用去离子水代替样品作为空白对照,并用Trolox溶液作为标准抗氧化剂组作标准曲线。根据标准曲线计算出样品的ABTS阳离子自由基清除能力。
1.4 数据统计分析

2 结果与分析
2.1 碱性蛋白酶控制酶解SPI规律探究
2.1.1 DH与表观浊度的变化
实验首先对SPI在0~24 h的酶解过程中蛋白DH及产物表观浊度的变化进行了探究。由图1可知,随着酶解时间的延长,DH整体呈上升趋势,其增长速率在4 h后逐渐降低并趋于平缓,与王利国、Xia Yichen等的研究结果较一致。此外,0~2 h酶解处理的样品表观浊度普遍较高,可能与蛋白胶体颗粒的存在有关,同时,样品浊度在酶解反应前期(0~1 h)急剧增加并在1 h达到最大,可能与碱性蛋白酶作用下蛋白疏水域的暴露有关,形成大量可溶性聚集体;随着酶解反应的进一步进行(4~24 h),样品因产生大量不溶性聚集体及亲水性多肽经离心变澄清。
采用SPSS17.0统计学软件进行数据处理,计量资料以表示,组间差异比较采用t检验,计数资料以%表示,组间差异比较采用χ2检验。
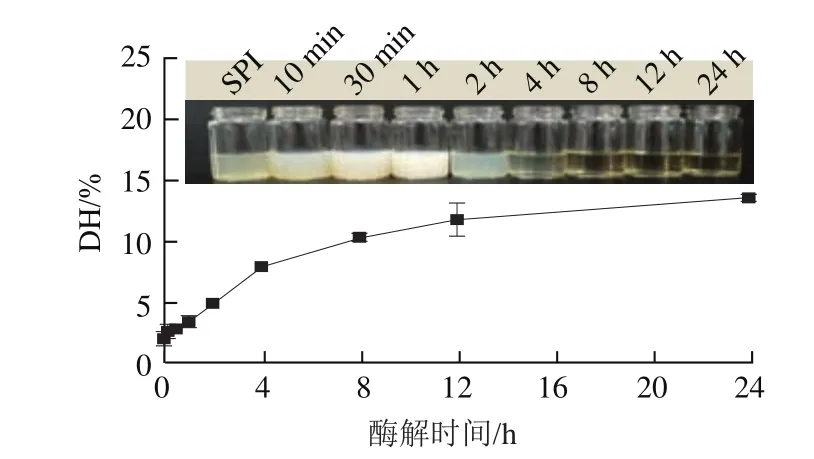
图1 DH与表观浊度随酶解时间的变化Fig.1 Effect of enzymatic hydrolysis time on DH and apparent turbidity of SPI
2.1.2 粒径、分散性、微观形貌及电位的变化
利用DLS技术对经碱性蛋白酶处理的蛋白样品进行水合半径粒径(-average size)和PDI的测定。一般认为,当PDI小于0.3时,所测定体系的均一性较好。由表1及图2可知,原始SPI粒径呈现多峰分布,且分散均一性较差(PDI>0.5)。实验发现控制酶解10 min~2 h(酶解1 h除外)结合热处理有利于SPI组装形成平均粒径在90~150 nm之间且分布较均一(PDI约0.2)的SPNs。结合表观浊度结果,酶解1 h样品未形成颗粒可能与大量可溶性聚集体的形成有关。而酶解4~24 h的样品粒径及PDI均较大。
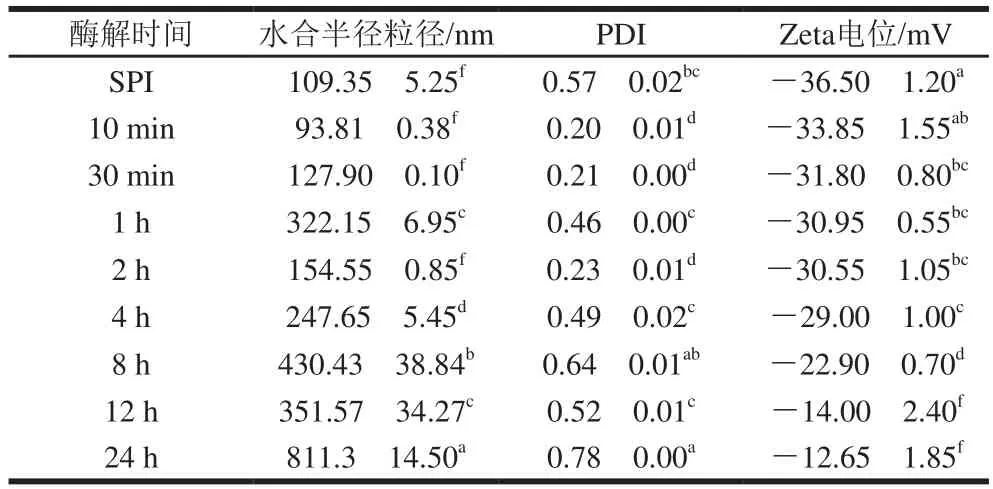
表1 SPI酶解产物的平均粒径、PDI和Zeta电位Table 1 Average particle sizes, PDI and zeta potential values of SPI hydrolysates
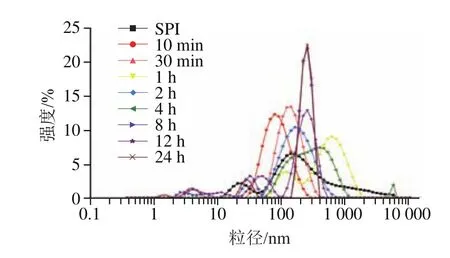
图2 SPI酶解产物的粒径分布Fig.2 Changes in particle size distribution of SPI hydrolysates as a function of hydrolysis time
进一步对SPI及酶解样品进行微观形貌观测,FESEM结果显示,原始SPI尺度较大且形貌不规则成(图3A);SPNs样品(图3B、C)均以大小均一的球形纳米颗粒存在,粒径较DLS结果小,可能与冷冻干燥处理造成的蛋白皱缩行为有关;而酶解4~24 h(以12 h为例,图3D)样品均为粒径较大的无规则聚集体,这可能与高DH下生成的无定形多肽有关。因此,基于DLS技术测定样品平均粒径对于酶解4~24 h样品可信度较低,暂不予以讨论。
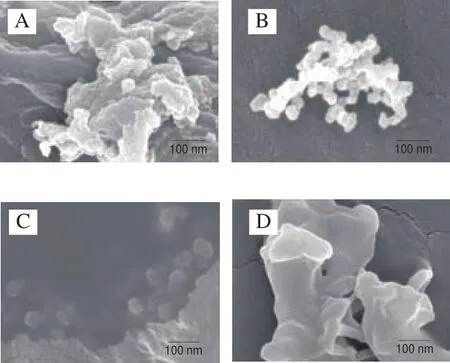
图3 SPI(A)及SPI经碱性蛋白酶酶解10 min(B)、2 h(C)及12 h(D)产物的FE-SEM图Fig.3 FE-SEM images of native SPI (A) and SPI hydrolyzed by alcalase for 10 min (B), 2 h (C) and 12 h (D)
随着酶解时间的延长,样品的Zeta电位值整体呈现出电位绝对值降低的趋势(表1),其中SPNs的Zeta电位绝对值均大于25 mV(30~35 mV),可以认为体系中有足够的静电斥力保持纳米悬浊液的稳定。而酶解4~24 h导致产物表面净电荷的显著下降,可能与酶解后期亲水性多肽的形成有关,而可溶性无定形多肽一般带电荷较分散且数量较少。以上这些结果表明,碱性蛋白酶酶解处理SPI可控制形成SPNs,DH是影响SPNs形成的关键因素。
2.1.3 SDS-PAGE分析
实验进一步对离心前后的酶解产物进行SDS-PAGE的对比分析,探究碱性蛋白酶作用下SPI亚基的降解及聚集规律。首先分析未经离心处理的酶解样品以确定各亚基在酶解过程中的降解情况,由图4可知,在非还原情况下,7S组分在酶解初期其、’亚基就发生了一定程度的降解,并很快降解完全。而亚基对碱性蛋白酶作用相对不敏感,整个酶解过程中亚基条带基本维持不变。11S组分中经二硫键作用形成的、亚基(约58 kDa),其条带经酶解处理后均消失,表明发生了新的聚集或者降解,同时随着酶解时间的延长,一条约35 kDa的新条带出现并在酶解4 h时开始逐渐加深。在还原情况下,酶解0~2 h样品有相对完整的亚基和亚基,表明在酶解及热作用下进一步发生了由二硫键诱导的聚集。酶解2 h后亚基开始降解,形成新的弥散条带,而亚基对酶解敏感性较低,结合非还原下SDS-PAGE的结果,亚基与亚基的降解片段可能通过二硫键作用形成了约35 kDa的新聚集体。
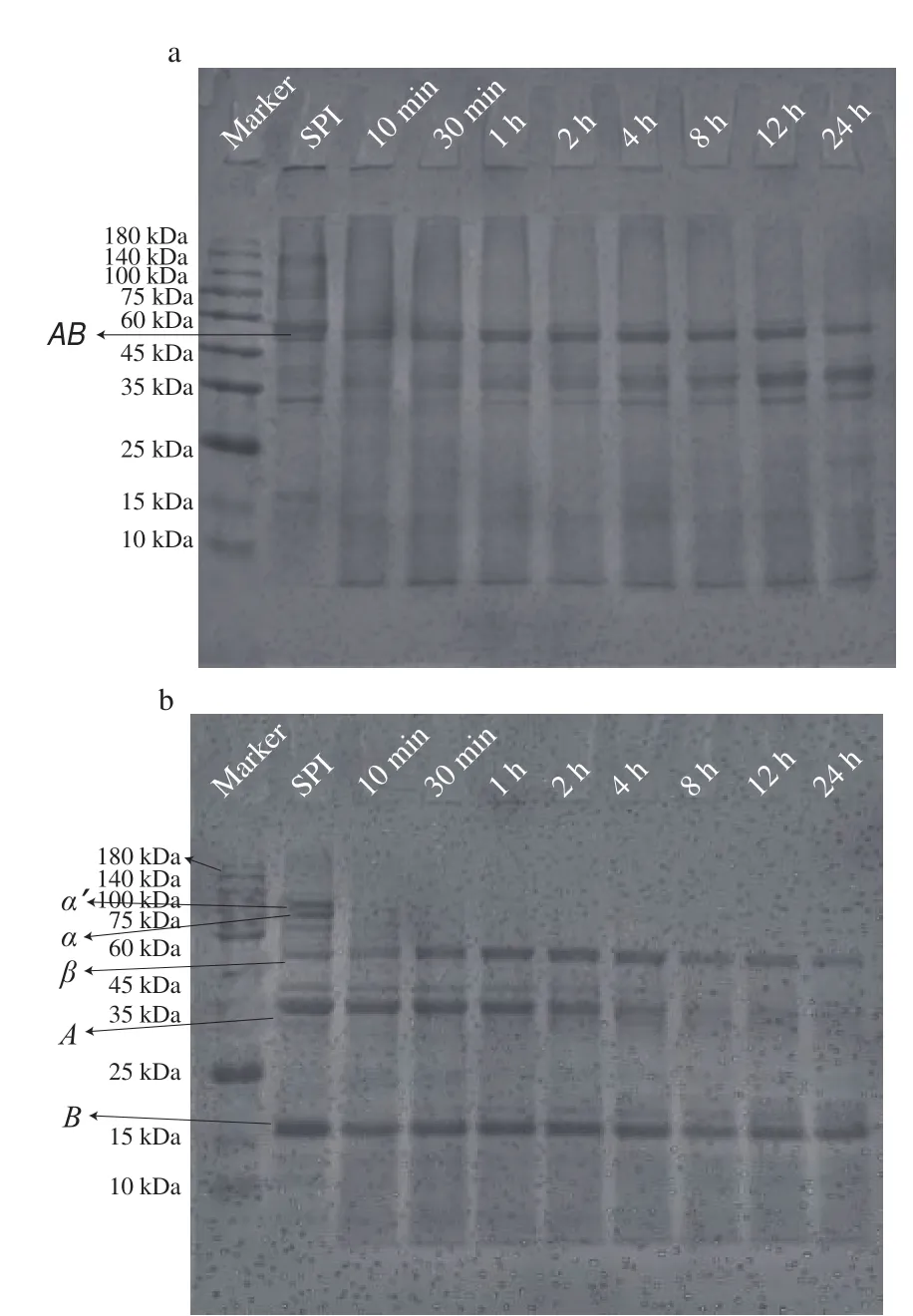
图4 经碱性蛋白酶酶解处理的SPI(离心前)电泳图谱Fig.4 SDS-PAGE pattern of SPI treated by alcalase(before centrifugation)
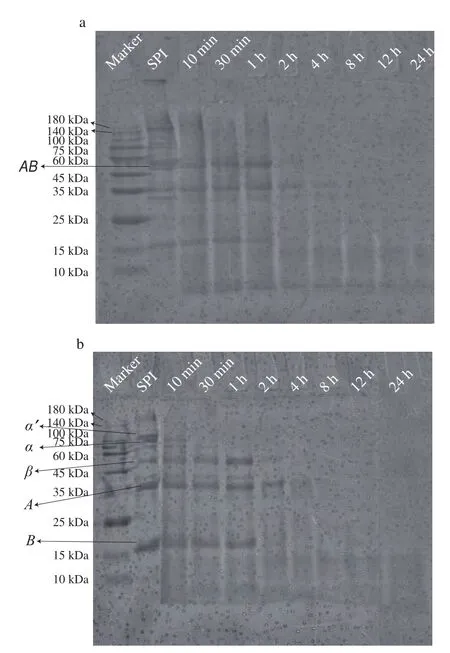
图5 经碱性蛋白酶酶解处理的SPI(离心后)电泳图谱Fig.5 SDS-PAGE pattern of SPI treated by alcalase (after centrifugation)
进而对经离心处理的酶解样品进行分析,实验发现酶解0~1 h的样品离心前后亚基变化不大(图4、5),其中酶解10~30 min的样品仅、’亚基发生部分降解,这有利于蛋白疏水区域的暴露及可溶性聚集体的形成,对应形成了亚基组成相对完整的SPNs(I类);而酶解1 h所对应的样品表观浊度最大(图1),表明形成最大尺度的可溶性聚集体,可能与与’的全降解相关。酶解2 h后,样品亚基消失且大部分亚基条带强度骤降,对应样品表观浊度下降,结合离心前亚基与亚基未出现明显降解的现象,可说明亚基的部分降解进一步促进蛋白疏水相互作用,并促使体系中可溶性聚集体向不溶性疏水聚集体转化,对应形成了以11S亚基及7S部分亚基为主导的SPNs(II类)。随着酶解的进一步进行,4~24 h酶解处理的样品离心后无特征亚基条带,表明样品主要以小分子肽组成,与其澄清表观对应(图1)。相关研究也发现大豆蛋白不同组分经水解处理后具有不同的聚集行为,Mohri等使用菠萝蛋白酶在低DH下对SPI及11S进行处理,发现11S的结构主导聚集行为,11S经热处理后结构展开,强疏水性的碱性亚基暴露于表面,而水解后的片段重新相互作用,将疏水性区域包裹在聚集物内部。上述结果表明,蛋白各组分与亚基的降解程度直接影响SPNs的组成与空间结构。
2.1.4 表面疏水性的变化
通过ANS法测定酶解过程中酶解产物的表面疏水性的变化,结果如图6所示,随着酶解时间的延长,酶解产物的表面疏水性表现出先增加后减少的趋势,酶解前期(10~30 min)增至最大(60 000~80 000),结合酶解过程亚基的降解情况分析,及’亚基的部分降解导致疏水区域的暴露,同时热变性会引起其结构的伸展并增强二硫键作用,二硫键周围常聚集着疏水性氨基酸,从而造成表面疏水性的增加;而随着酶解时间的延长(1~2 h),及亚基发生疏水聚集,表面疏水区域减少,表面疏水性相对酶解前期有所降低,所以在酶解的不同阶段所形成的两类SPNs具有不同的表面性质;酶解后期(4~24 h)显著降低,可能是此时体系中亲水性多肽占据主导地位,提高了体系的亲水性,同时由于肽键裂解引起的分子电荷增加导致疏水性(可检测的)降低,-螺旋与-折叠结构域之间的疏水结合位点丢失也会造成疏水性降低,这与其他类似酶解研究中结果一致。
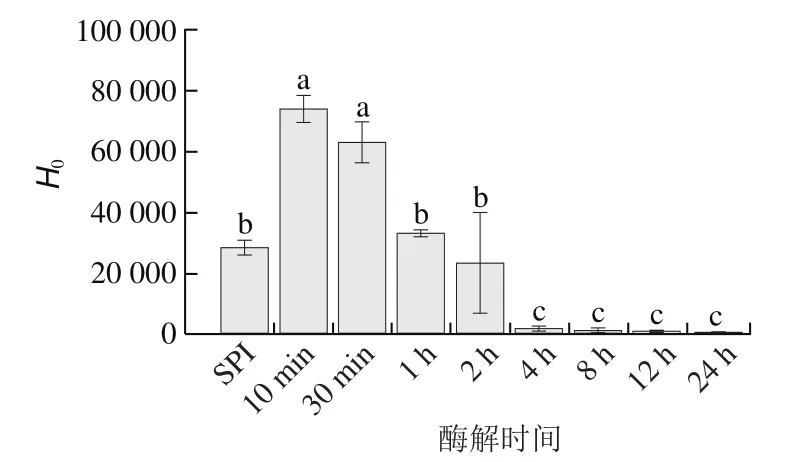
图6 SPI酶解产物的表面疏水性Fig.6 Surface hydrophobicity of SPI hydrolysates
根据以上实验结果,将酶解过程划分成低DH(0%~4%)、中DH(4%~8%)、高DH(8%~12%)3 个阶段以便进行后续颗粒形成机制的相关研究,其中低DH阶段形成I类颗粒具有高表面疏水性的SPNs-DH 3%,中DH阶段形成II类颗粒亲水性更高的SPNs-DH 5%,高DH阶段未形成有序的颗粒结构。
2.2 SPNs形成机制探究
2.2.1 CAC分析
芘作为一种强疏水荧光探针,在335 nm激发后其荧光发射光谱有5 个峰,峰1(373 nm)的荧光强度和峰3(385 nm)的荧光强度之比(/)强烈地依赖于其周围微环境的极性,/值越低则反映芘所处微环境的疏水性越高。从图7可以看出,当蛋白质量浓度很低时(小于CAC),/值基本不变,这是因为此时蛋白分子以单体或无规则结构存在于水溶液中,其质量浓度的增加对分散于溶液中的芘所处环境的影响不大。随着蛋白质量浓度的增加,/逐渐降低,通过Boltzmann拟合发现,原始SPI的CAC为0.25 mg/mL,低DH阶段SPNs-DH 3%的CAC仅为0.09 mg/mL,此时亚基疏水结构的暴露导致表面疏水性更高(图6),在较低蛋白质量浓度下即可发生聚集形成颗粒结构;中DH阶段II类颗粒SPNs-DH 5%的CAC相对I类颗粒SPNs-DH 3%略有增加(0.25 mg/mL),可能与其表面疏水性的下降有关(图6);而高DH阶段的CAC显著增加至11.56 mg/mL,结合表面疏水性的结果也说明体系中亲水性结构占据主导地位,疏水相互作用不足以形成有序的纳微结构。
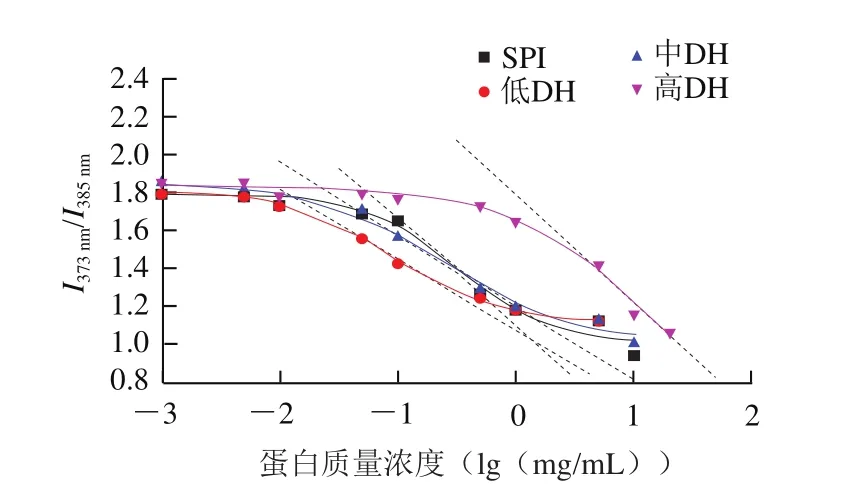
图7 SPI及低、中、高DH酶解样品的芘发射荧光强度比值(I373 nm/I385 nm)随蛋白质量浓度变化的Boltzmann拟合函数Fig.7 Boltzmann curves of pyrene fluorescence intensity ratio I373 nm/I385 nm of SPI and hydrolyzed SPI with different DH levels (low, middle and high)against logarithmic protein concentration
2.2.2 二级结构的变化
通过CD分析酶解样品的二级结构,在碱性蛋白酶作用下,低DH、中DH、高DH下酶解产物的CD图谱随DH的增加而明显往短波长方向移动(图8),同时负峰逐渐收缩加深;相应地,其-折叠含量随DH的增加不断上升,-螺旋和无规卷曲含量相应减少(表2),表明-螺旋和无规卷曲向-折叠结构的转变,这可能与其、’亚基的降解有关,同时还导致了其-螺旋/-折叠比例的明显下降,表明其结构柔顺性在酶解过程中不断上升。上述研究结果表明,-螺旋和无规卷曲向-折叠结构的转变有利于SPNs的形成,并且,与II类颗粒SPNs-DH 5%相比,I类颗粒SPNs-DH 3%的结构刚性更大。

图8 SPI及低、中、高DH酶解样品的CD图Fig.8 CD spectra of SPI and hydrolyzed SPI at different DH levels(low, middle and high)
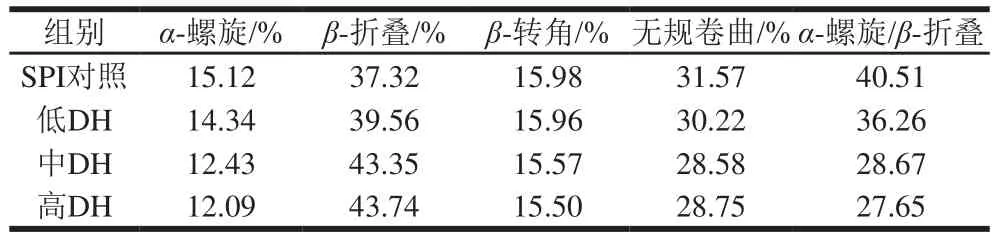
表2 SPI及低、中、高DH酶解样品的二级结构组成Table 2 Secondary structure contents of SPI and hydrolyzed SPI at different DH levels (low, middle and high)
2.2.3 相互作用力
一般认为DTT、SDS和尿素可分别破坏蛋白质的二硫键、疏水相互作用力以及氢键。如图9所示,经各变性剂处理后,整体而言低DH阶段形成的I类颗粒SPNs-DH 3%与中DH阶段形成的II类颗粒SPNs-DH 5%的表观浊度与粒径变化规律相似,说明维持颗粒结构的整体作用力类似。当SDS单独作用时,两类颗粒的粒径均显著减小,说明颗粒的整体结构由疏水相互作用力维持。在SDS与DTT共同作用下,颗粒粒径均比单独SDS处理小,当SDS将SPNs解聚后,暴露的内部结构在DTT的作用下进一步被打开,说明二硫键主要参与稳定颗粒的内部结构。尿素单独作用时,粒径略微减小,溶液浊度稍有减小,说明氢键主要位于颗粒外部,氢键的破坏导致外部结构的部分解离;对比两类颗粒粒径变化情况,当DTT与尿素两者共同作用时,SPNs-DH 3%的粒径无明显变化,而SPNs-DH 5%粒径显著增加,说明氢键与二硫键同时被破坏时,SPNs-DH 5%中内部所含的折叠结构发生了展开。
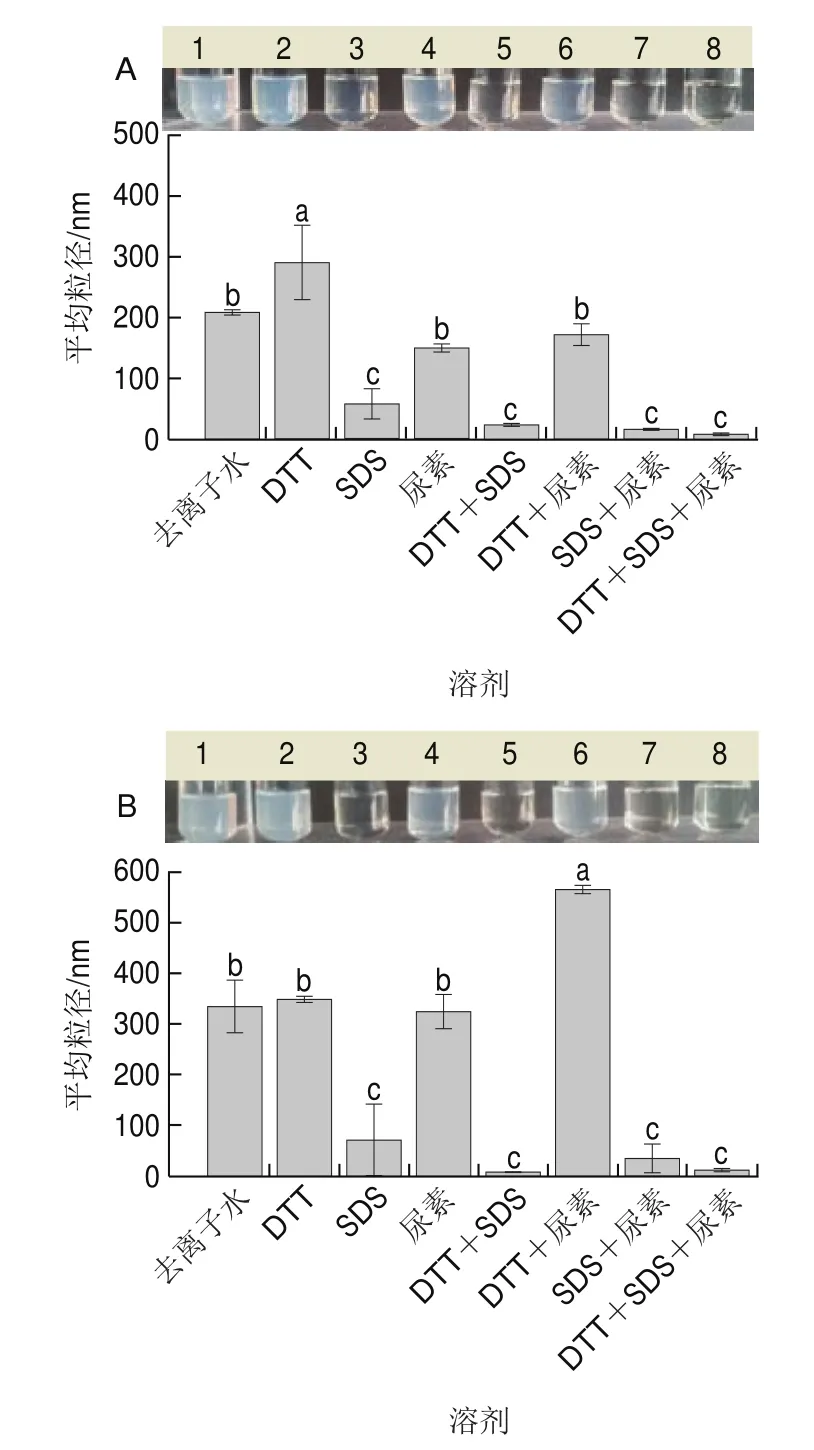
图9 SPNs在不同蛋白变性溶剂中的外观和粒径Fig.9 Appearance and average particle size of SPNs in different protein-denaturing solvents
根据以上实验结果,推测的SPNs形成途径模型如图10所示,低DH下,亚基与亚基通过二硫键与疏水相互作用发生聚集,形成了疏水核心。亚基与亚基之间通过二硫键作用,形成了颗粒的中间结构。同时低程度酶解使得相对更亲水的、’亚基暴露,并包裹于颗粒的表面,在氢键的作用下形成了颗粒的最外层结构,另外由于、’亚基具有延伸区域可抑制SPI的聚集,使得I类颗粒SPNs-DH 3%能够维持均一分布;中DH下,、’亚基完全降解,亚基被暴露出来并且部分降解,原本由亚基与亚基组成的疏水内核发生结构的展开,仅部分亚基可再次组成疏水内核,形成了以部分亚基及两亲性肽为主要表面结构的II类颗粒SPNs-DH 5%;高DH下,由于亚基的持续降解与亚基发生彻底的疏水聚集,内部疏水相互作用不足以使体系中存在的酶解片段形成颗粒结构。
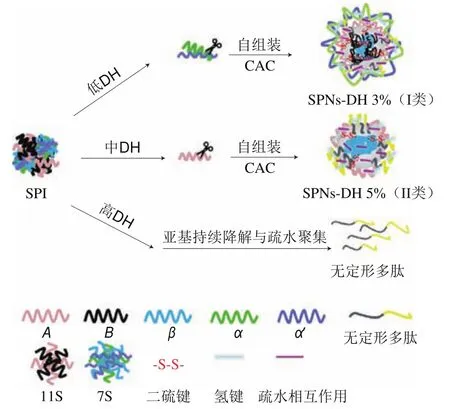
图10 SPNs-DH 3%及SPNs-DH 5%的形成机理图Fig.10 Schematic illustration of the formation mechanism of SPNs-DH 3% and SPNs-DH 5%
2.3 抗氧化活性的评价
如图11所示,经过不同程度酶解后的产物,其抗氧化性较于原始SPI均有所提升,且随着DH变大有增加的趋势,结合酶解过程中蛋白亚基组成的变化规律,说明酶解过程中、’亚基以及亚基降解生成的抗氧化性多肽提升了体系的抗氧化性。
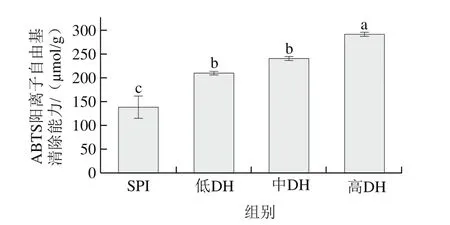
图11 酶解前后ABTS阳离子自由基清除能力的变化Fig.11 Changes in ABTS radical cation scavenging activity of SPI before and after enzymatic hydrolysis
3 结 论
以SPI为原料,利用碱性蛋白酶对其进行控制酶解处理,成功制备了SPNs-DH 3%与SPNs-DH 5%两类分布均匀(PDI<0.3)、粒径可控(90~200 nm)的SPNs,其中DH及亚基解离/降解是颗粒形成的关键性因素。I类颗粒SPNs-DH 3%包含相对完整的7S及11S亚基,表面疏水性较原始SPI有显著提升,且在更低质量浓度下可自组装形成均一的聚集体,由疏水相互作用维持颗粒的整体结构,氢键与二硫键维持表面结构与内部结构;II类颗粒SPNs-DH 5%是由亚基及部分亚基主导的亲水性胶体颗粒,与I类颗粒SPNs-DH 3%相比,II类颗粒SPNs-DH 5%中形成更多由氢键与二硫键稳定的折叠结构,而表面疏水性略有下降,CAC略有增加。由于酶解过程中释放了抗氧化肽段,与SPI相比,所形成的两类SPNs抗氧化性均有显著提升。综上,利用碱性蛋白酶控制酶解SPI可形成不同表面特性的抗氧化蛋白纳米颗粒,为生物酶解构建蛋白基颗粒,进一步提升蛋白功能特性与扩大应用范围提供新方案。
