大数据时代土壤微生物地理学研究综述
2022-07-30靳一丹陆雅海
靳一丹,陆雅海
北京大学,城市与环境学院,北京 100871
生物地理学是一门旨在记录和理解生物多样性空间格局的科学,其目的是揭示生物的丰度和组成的产生和维持机制[1—3]。几个世纪以来,生物学家一直在探索大型生物的生物地理分布格局[4—5];直到1934年,荷兰学者Baas-Becking提出了著名观点“万物无所不在,但由环境选择”(Everything is everywhere: but the environment selects),首次将微生物与自然环境联系起来,推动了微生物生物地理学的发展[6]。“微生物”通常指细菌域、古菌域的成员和真核生物域的微小成员,它们在地球上数量极多、分布广泛,是地球生物多样性的重要组成部分[1,7]。土壤含有地球上最多样的微生物组,它们在物质分解、元素生物地球化学循环、植物生产力和生物健康中扮演着重要角色[8—11]。理解形成土壤微生物生物地理分布格局的原因及其可能产生的结果,不仅有助于理解物种进化过程(如物种形成)和生态过程(如物种演替、群落发展、物种的传播和维持),还可以为调节生物多样性和生态系统复杂性的机制提供重要信息[12—14]。
受到传统观察手段的技术限制,许多早期研究认为土壤微生物多样性分布的空间变化是“背景噪音”[4]。随后出现的分子生物学技术提高了环境微生物识别的速度和准确性,这类技术与大数据分析的耦合大大推动了土壤微生物生物地理学的发展[15—16]。国际上,中国和美国对近代土壤微生物生物地理学研究的贡献度最大,其中土壤细菌最受关注;我国的相关研究正在逐年增加,其研究方向主要为农业、生态与环境科学(图1)。由此可见,土壤微生物生物地理学在国际上和我国国内都处于广受关注且快速发展的阶段。本文首先阐述土壤微生物生物地理学的研究进展,接下来重点介绍分子生物学技术和大数据分析的应用,并对土壤微生物生物地理学未来的发展方向进行展望。
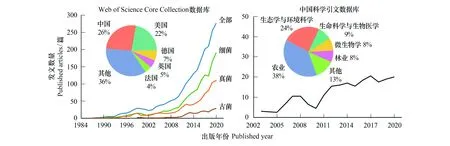
图1 国际期刊和我国出版期刊的发文数量随时间的变化Fig.1 The number of published articles across time in international journals and in Chinese journals
1 土壤微生物生物地理学研究进展
1.1 土壤微生物地理分布格局的发现及其与大型生物之间的异同
土壤微生物地理分布格局的探索经历了“从否定到肯定”的过程。在21世纪前,依赖于显微镜和富集培养技术,科学家们发现一些原生生物具有世界性分布的特点[5,17]。21世纪后,在纯培养和分子生物学技术的帮助下,人们发现土壤微生物也具有地方性特点并呈现出地理分布格局[10,18]。分子生物学技术提高了环境微生物的识别分辨率,这些微生物表现出的生物地理格局否定了以往认为土壤微生物群落是一个没有空间结构的“黑匣子”观点。微生物的生物地理学概念晚于动植物两个世纪,判断两者是否存在共通模式和原理将有助于在现有的宏观生物地理学体系下发展适用于土壤微生物的理论框架,且微生物地理研究也可对现有的生物地理学理论进行补充和验证[2,6]。土壤微生物和大型生物(特别是植物)常常密切相关,我们可以合理推测微生物与大型生物的生物地理存在一定关联[1,19]。相关研究进展包括以下三方面。①从生物的分布格局来看:土壤古菌和真菌的多样性与许多大型生物表现出一致的趋势,且真菌的全球分布格局还符合动植物的Rapoport法则[10—11]。这可能是由于土壤真菌与植物之间具有更密切的联系(如共生关系),导致其地理分布受到植物分布的制约[10]。②从生物的生态模式来看:常被用于大型生物生态学研究的“种-面积关系”和“共现模式”其实也适用于微生物[9,20]。③从造成生物分布格局的原因来看:研究发现,一些驱动微生物多样性模式的因素可能与解释大型生物多样性模式的一些基本过程类似[21—22]。由此可见,土壤微生物与大型生物在生物地理分布上有一些共同点。
然而,仍有一些研究展示了土壤微生物(主要是细菌)与大型生物在地理分布上的不同。Bahram等人通过全球表土微生物组调查发现,土壤细菌的分类多样性和基因功能多样性都不符合大型生物普遍的生物多样性纬度梯度格局[11]。除了纬度梯度,一些土壤微生物的多样性在海拔、降水梯度和不同土地利用类型上的分布特点也与大型生物不同[23—25]。微生物的微小体型、单细胞特点、繁殖方式和生理特征都与大型生物有明显的不同;且由于生存环境的差异,某些土壤微生物的生物地理分布主要由土壤特性控制[26]。这些都可能导致土壤微生物生物地理分布不同于大型生物。
由此可见,土壤微生物与大型生物的生物地理格局不完全一致,需要发展出更具包容性的生物地理学概念和理论来解释这些差异。为了完成这个目标,不仅需要考察土壤微生物的分布和多样性,更重要的是将微生物信息与环境信息联系起来,从而探索产生土壤微生物地理分布格局的内在机制。
1.2 土壤微生物生物地理分布格局的驱动因素
传统生物地理学和微生物生物地理学都认为当代环境因素(包括生物和非生物因素)和历史事件(包括扩散限制和过去的环境条件)是研究生物分布格局的两大主题[1,27]。基于以上两个主题,Martiny等人构建了一个微生物生物地理学框架,其中包括四个假说:①微生物在空间中随机分布;②微生物的生物地理学反映了当代环境变化的影响;③所有的空间变化都来自于历史事件的持续影响;④微生物的分布是历史事件和当代环境共同影响的结果[1]。基于以上框架,研究者们就导致土壤微生物分布格局的两大主题进行了探索。
1.2.1当代环境因素
非生物因素包括气候和土壤特性,其中土壤特性对土壤微生物的组成和分布起着至关重要的作用[28—29]。土壤pH是一个有效的生境“筛选器”:研究发现土壤pH与某些微生物类群的丰度密切相关,且被认为是土壤细菌群落的最佳预测因子[25,30—31]。除了pH,土壤养分变化也会在不同程度上影响土壤微生物的群落组成和多样性[28]。除了土壤特性,气候(如气温和降水)则可能是通过改变土壤分解速率,从而间接地影响土壤微生物的地理分布[32]。
生物因素包括微生物本身的特征和环境中其他生物的影响。微生物的定殖、物种形成和灭绝都直接受到其生活史特征的影响,它们庞大的种群规模和快速增长的特点为遗传多样性提供了巨大的潜力[7]。土壤中不同生物群落(包括微生物和较大体积的生物)间存在相互作用,例如:真菌紧密联系的群落特点可能导致它们具有更强的环境变化抵抗能力[33];植物根系分泌物和凋落物不仅改变土壤养分条件,而且深刻影响土壤微生物与植物之间的共生关系。这些都是影响土壤微生物分布格局的生物因素[34]。
1.2.2历史事件
扩散为生物个体(及其所代表的类群)被动或主动地从一个地点移动到另一个地点并成功定居的过程[3]。长远的扩散距离、不利环境条件以及地理屏障等都会导致不同地理位置微生物组成和多样性的差异[35,36]。历史事件和当代环境因素并不相互排斥,它们共同驱动形成土壤微生物群落的空间格局,但它们的相对重要性仍存争议,需要进行更系统化的比较[18,27,37]。
1.3 土壤微生物群落的构建规则
基于当代环境因素和历史事件的微生物生物地理学框架有助于解释土壤微生物的地理分布格局,而探索微生物群落的构建规则有助于深入理解土壤微生物群落结构的形成和演替。目前认为微生物群落的构建受到确定性过程和随机性过程这两种基本生态过程的影响[38]:在确定性过程中,物种受到非生物和生物因素的选择作用,将具有其独特的生态位,能通过竞争生存下来;而在随机性过程中,则涉及物种随机的出生、死亡、扩散和遗传漂变事件,许多物种存在于相同或重叠的生态位中,但由于它们的竞争能力处于紧密平衡状态而不会相互淘汰。
实际上,确定性过程和随机性过程同时影响土壤微生物群落的构建,它们的相对重要性则会因观察角度的不同而产生差异[39—40]。从空间尺度上看,随机性过程可能对较小范围内土壤细菌群落的构建起主导作用;但在较大范围内,起主导作用的则是确定性过程[38]。从时间尺度上看,细菌群落的构建在土壤演替早期受随机性过程的控制;随着演替的进行,确定性过程的相对重要性逐渐增加[40—41]。除了观察的时空差异,微生物个体大小也会使观察结果发生变化[14]。基于以上差异,研究者们构建了各种概念模型[14,40,42],用于系统地了解生态系统中微生物群落的形成和演替特征(图2)。

图2 微生物群落构建概念模型Fig.2 Conceptual model of microbial community assembly图片根据Dini-Andreote等人[40]、Wang等人 [42]和Luan等人 [14]的研究
2 分子生物学技术推动土壤微生物生物地理学快速发展
2.1 分子生物学技术对土壤微生物生物地理研究的重要性
分子生物学技术以具有特异性的DNA片段为研究对象,可以检测环境微生物的丰度和多样性[43—44]。自1998年宏基因组学概念的提出,以核酸为研究对象的分子生物学技术被广泛应用于环境微生物研究中[45]。与传统微生物研究方法相比,结合生物信息学的现代分子生物学技术分辨率更高,能不依赖培养技术而直接获得自然环境中的微生物信息,既可以为纯培养条件(如培养基成分、培养温度等)的优化提供帮助,也可以从分子水平上判断环境微生物的丰度和多样性。

图3 基于已有数据库(绿色流程)和采样(棕色流程)的土壤微生物生物地理学研究示意图Fig.3 Diagram of the researches in soil microbial biogeography based on existing databases (green pipelines) or on new studies (brown pipelines)其中一些图片根据Thompson等人[46]、Mardis等人[47]、van den Hoogen等人[48]、Ma等人[15]和Garcia-Pichel等人[49]的研究
在分子生物学技术研究初期,主要用于土壤微生物生地理研究的是基因克隆文库和基因指纹图谱技术,然而这些技术具有成本较高、工作量大、耗时长的缺陷,且不能提供微生物数量和基因表达水平方面的信息,因此不适合用于观察和监测复杂环境中微生物多样性的时空动态变化[30,45]。与之相比,随后出现的高通量测序技术,一次能对几十万至几百万条DNA分子进行序列测定,具有速度快、成本低的优点[50]。基于这些优点,高通量测序目前已成为研究各种尺度下土壤微生物生物多样性、地理分布格局和群落构建演化机制的首选技术(图3)。
2.2 高通量测序(High-throughput sequencing)平台
高通量测序平台的代表是Roche公司的454测序仪、Illumina公司的Solexa测序仪和Applied Biosystem公司的SOLiD测序仪[51]。以下简要介绍目前应用最广的Roche 454测序平台和Illumina测序平台。
2.2.1Roche 454测序平台
Roche 454平台利用微乳液PCR和焦磷酸测序技术确定核酸的碱基序列,具有测序读长较长(最长1000 bp)、耗时短的优点[52]。通过识别小亚基rRNA基因序列的差异,454焦磷酸测序技术被广泛应用于土壤微生物生物地理研究中,包括微生物多样性分析、探索微生物地理格局、构建微生物预测模型等[30,53]。然而,Roche 454测序平台存在通量相对较低、试剂成本高、重复碱基聚合区域测序错误率高的问题,这些缺陷限制了焦磷酸测序被应用于大样本量的研究中[52]。
2.2.2Illumina测序平台
Illumina平台利用桥式扩增和合成测序技术确定核酸的碱基序列,是目前高通量测序应用最广泛的平台,具有通量高、低成本的优点[52]。Illumina公司包括HiSeq、MiSeq、NextSeq、NovaSeq等平台,前两个平台的使用较多。这些平台的主要区别在于总输出和最大读取长度,如较新的HiSeq 4000系统一次运行的通量可达1.5 Tb,但读取长度仅限于150 bp;而MiSeq系统的最大读取长度为500—550 bp,但输出量受限(15Gb)[54—55]。可综合考虑研究目的、通量要求、测序成本后再选择合适的Illumina平台。得益于Illumina测序平台通量高、成本较低的特点,全球尺度的土壤细菌(16S rRNA基因测序)、古菌(16S rRNA基因测序)、真菌(ITS基因测序)多样性调查得以实现[19,49]。
3 大数据分析实现了多样品与大尺度的微生物生物地理研究
3.1 大型国际微生物计划
随着基因测序领域的快速发展,人类对微生物多样性及其功能有了新的认知。然而,由于不同微生物组的研究之间缺乏合作,“碎片化”的测序数据和背景数据缺乏标准分析方法,使大规模微生物组的研究受到限制。为了解决这一问题,各种国际微生物计划自21世纪以来被相继提出,如海洋微生物普查计划(International Census of Marine Microbes, ICoMM)、人类微生物组计划(Human Microbiome Project, HMP)[56]、地球微生物组计划(Earth Microbiome Project, EMP)[57]、全球土壤生物多样性倡议(Global Soil Biodiversity Initiative, GSBI)等,其中EMP和GSBI大大推动了土壤微生物多样性研究领域的发展。
3.1.1地球微生物组计划(EMP)
EMP(http://www.earthmicrobiome.org)成立于2010年,其目的是通过对地球微生物群落的大规模取样,得到全球未培养微生物多样性目录(包括样本信息及其元数据),并使用标准化流程来探索影响微生物群落结构的生物地理学原理[57]。在全球五百多名科学家的共同努力下, EMP在2017年发表了他们的第一个荟萃分析成果,包括新建立的环境微生物基因序列参考数据库,并提出了整合不同研究数据的新思路,将微生物多样性的探索推向前所未有的规模[46]。该成果已被广泛应用于各类环境微生物研究中:如Zhang等人通过比较EMP发布的序列和来自公共数据库的基因组数据,揭示了地球生物群落原核基因组测序的现状[16];Ma等人则通过提取EMP数据库中14种环境的大量测序信息,绘制了地球微生物的共存网络[15];而Luan等人则利用EMP的测序数据和分析平台,揭示了微生物群落构建与生物有机体大小之间的关系[14]。除了以上研究,EMP的数据也大大推动了测序数据分析工具、分析平台的开发和测试以及模型构建的进程[58—59]。
3.1.2全球土壤生物多样性倡议(GSBI)
GSBI(www.globalsoilbiodiversity.org)发起于2011年,其目标是通过全球科学家的合作,整合跨学科成果,向公众提供信息并推广到环境政策中,为当前和未来土壤可持续性研究创建平台。与EMP不同的是,GSBI旨在更好地利用已有的土壤生物多样性和生态系统服务方面的知识,而不是开始新的研究。至今,全球性的土壤微生物研究的对象包括细菌[60]、真菌[10]和原生生物[48],这些研究成果对绘制全球土壤微生物地图集、探寻驱动多样性和地理分布格局因素、研究群落构建机制和构建预测模型具有重大贡献。
3.2 微生物序列数据库和测序数据分析平台的兴起
不断推进的大型国际微生物计划使全球环境微生物数据量呈爆炸式增长,随之产生了各种微生物序列数据库,其中信息的挖掘离不开生物信息学和计算机科学的支持[61—62]。为了满足生命科学日益增长的计算需求,各类微生物测序数据分析平台快速兴起,成为了不具备专业生物信息学和编程技能的研究人员深入分析大型数据集的有力工具,大大节约了数据分析和结果可视化的时间(图3)。以下是一些近期兴起的环境微生物序列数据库和测序数据分析平台以及它们在微生物研究中的应用。
3.2.1全球真菌数据库(GlobalFungi)
GlobalFungi数据库(https://globalfungi.com/)是目前最全面的全球真菌数据平台,包含所有陆地生境中的真菌群落信息,且具有网页交互界面实现查询结果的可视化[63]。GlobalFungi数据库于2021年5月1日发布了最新版本3(Release 3),包括超过十一亿的真菌内转录间隔区(ITS)序列和两亿多个ITS序列变体,这些序列来自于367个研究的三万多个具有地理信息和元数据的样品。GlobalFungi数据库既可以被用于全球真菌的生态与地理研究,也可以作为特定真菌类群的辅助研究工具[64—65]。该数据库还允许作者提交尚未涵盖的研究数据,并以这种方式为真菌系统学、生物地理学和生态学提供资源。
3.2.2陆地宏基因组数据库(TerrestrialMetagenomeDB)
TerrestrialMetagenomeDB数据库(https://webapp.ufz.de/tmdb/)建立于2020年,是第一个专门的陆地宏基因组数据库,不仅包含全球范围各种生物群落和材料的宏基因组测序数据,还包含生物样品的元数据和测序技术的元数据[66]。TerrestrialMetagenomeDB数据库目前已更新到2.0版本,包括来自SRA(Sequence Read Archive)和MG-RAST(Metagenome Rapid Annotation using Subsystem Technology)的两万多个陆地宏基因组,并对它们的数据集进行了内容补充和格式统一。该数据库的特点是具有用户友好型界面,既可以使用关键词搜索,也可以直接从地图上按照地点快速选取感兴趣的宏基因信息,用于后续新旧数据的融合分析或荟萃分析。
3.2.3原核生物信息数据库及生境预测平台(ProkAtlas)
虽然原核生物在全球各种生境中广泛分布,但它们有各自的环境偏好,从而形成全球微生物的多样性格局。在这个背景下,2020年出现了一款名为ProkAtlas(https://msk33.github.io/prokatlas.html)的原核生物信息数据库,可以通过网页交互作业对给定原核生物群落的生境进行预测[67]。为了避免扩增子测序引物引起的偏差,ProkAtlas数据库使用的是从五千多个宏基因组项目中提取的三十多万个16S rRNA基因序列,其主要来源为根际、土壤和海洋等环境样品。利用这些宏基因组项目的元数据,可以将基因与生境进行关联,从而对“微生物群落适应的环境类型”、“微生物的生境广谱性”以及“该微生物祖先或后代的居住生境范围”等问题进行解答。
3.2.4微生物组学分析和可视化平台(Anvi′o)
Anvi′o(Analysis and visualisation platform for ‘omics data)是一个开源的、先进的微生物组学分析和可视化平台,主要用于泛基因组分析,也适用于宏基因组学、宏转录组学和系统基因组学及其综合分析。该平台包含一百多个可单独执行任务的程序,具有灵活性、交互性和模块化的特点,满足用户构建分析工作流程的需求[68]。在基因组和泛基因组分析流程中,Anvi′o可以识别并区分密切相关的微生物基因组中的核心基因和附属基因;而在宏基因组分析流程中,Anvi′o可以获得微生物的群落组成并完成可视化[69—70]。除了‘以上完整的工作流程,Anvi′o的各个模块还可以被单独使用:如进行宏基因组分箱、基因组精炼、基因组完整性考察等。得益于30多位开发者的贡献,Anvi′o平台目前拥有超过八万行Python和JavaScript代码,用于满足微生物学家复杂的需求。
3.2.5微生物组学数据平台(MicrobiomeAnalyst)
MicrobiomeAnalyst(https://www.microbiomeanalyst.ca/)是一个微生物组学数据的分析网站,目前具有标记基因数据分析、鸟枪数据分析、公共数据投影和分类集群富集分析四个模块,即使不具备专业编程技能的人员也可以利用该平台对微生物组学数据进行预处理、统计分析、功能分析、荟萃分析和可视化[71]。目前,MicrobiomeAnalyst平台已被广泛运用于自然环境(如土壤、沉积物、海洋)微生物和肠道微生物的研究中[72—75]。
3.2.6在线微生物分析平台(Qiita)
Qiita(https://qiita.ucsd.edu/)是一款开源、简便、快速的在线微生物分析平台,既包括QIIME等测序数据的分析流程,也具有面向全球的公开数据库,可以帮助用户对多个具有多组学数据的研究进行跟踪,适合初学者使用[76]。Qiita不仅拥有超过十万个扩增子数据集和超过五百个配对的宏基因组和代谢组学数据集,还具有标准化的技术、数据和分析工具,提供了一个大规模数据整合研究的框架,减轻了微生物生态研究者的技术负担[77]。Qiita平台允许使用最新的分析技术快速重新分析已存储的数据集,即使是非生物信息学家也能够使用标准化流程轻松地进行数据分析和荟萃分析。除了土壤微生物生物地理学研究[78],Qiita在环境污染、人类疾病与健康、分子生物学方法探索中也起到重要作用[79—81]。
3.2.7宏基因组全球目录平台(gcMeta)
宏基因组全球目录(Global Catalogue of Metagenomics, gcMeta)平台是中国科学院微生物组学研究计划(CAS-CMI)的一部分,也是世界微生物数据中心(WDCM)的合作数据库[82]。gcMeta(https://gcmeta.wdcm.org/)平台具有自己的数据库,包括四万多个宏基因组数据、九万多个扩增子数据、一千多个宏转录组数据和三千多个基因组数据,涵盖人类肠道、宿主微生物、土壤等诸多研究领域;除此之外,gcMeta也提供网页版的数据分析工具(如组装、注释、可视化等)和工作流程(如基因组注释、宏基因组分类等)。未来,gcMeta平台有望对与人类疾病与健康有关的微生物的研究起到重要贡献[83—84]。
4 展望
4.1 微生物分类分辨率的统一与优化
在大多数微生物测序数据的分析中,微生物类群的划分依赖于一个或多个基因组区段的核苷酸序列的相似性(即聚类法),这种人为设置相似度阈值的聚类方法会使微生物类群因分类分辨率的高低而变化,这种灵活性使得解释生物地理格局的过程更加复杂[3]。针对这类问题,有研究者在土壤微生物生物地理研究中选择使用扩增子序列变体(Amplicon Sequence Variants, ASVs)——而不是使用传统的聚类结果(Operational Taxonomic Units, OTUs)——对微生物进行分类[15,46]。ASVs是基于测序区段序列差异的分类结果,可以作为一种稳定的分类标识符,与OTUs相比具有可重复使用的优势,即使在未来也可以与16S rRNA基因或基因组数据库进行比较[46,85]。因此,未来建议使用ASVs代替传统的OTUs聚类方式来对微生物进行分类,或是针对研究目的对微生物聚类分类系统的分辨率进行统一与优化,一方面有利于来源于不同数据库信息的整合,另一方面可以避免由于分类分辨率过低而低估了微生物的地理分布特点。
4.2 利用微生物生物地理学来验证或构建生态模型
随着大型国际微生物计划的不断推进,全球土壤微生物数据量呈爆炸式增长,各种新兴数据库和数据分析平台的推广有助于各类微生物系统模型的构建,这些模型将对验证生态学猜想和预测土壤生态变化起到重要作用。
与大型生物相比,以微生物作为观察对象的优点之一是其可以被用于进行可控且可重复的试验。首先,微生物培养试验和生理生化试验为直接检验某些生态学猜想(特别是那些野外无法检验的大型生物的猜想)提供了可能性,从而帮助人们更深入地理解生物多样性的形成过程[49,86]。其次,通过微生物生物地理研究和试验验证而构建的微生物系统模型可以成为研究生态学的有用工具[59]。至今,全球尺度下的土壤微生物研究不仅包括绘制各类土壤微生物的全球分布图谱[60,87],还包括微生物组内的营养结构调查[88],这些研究将对全球环境变化导致的土壤生态变化的预测起到重要作用[89]。在未来有关土壤微生物模型构建的研究中,一方面可以结合来自EMP、GSBI等计划发布的测序数据,综合考虑各种环境背景;另一方面还需关注土壤、气候以及其他生物与微生物之间的相互关系,构建更精确的土壤微生物预测模型。需要注意的是,数据量增大的同时构建模型的噪声也会增加,结果可能导致模型准确性不增反减[90]。对于目前指数增长且来源多样的各类环境微生物测序数据,需要选择合适的去噪手段(如平滑处理、分类处理等)对它们进行前处理后再增大模型输入的数据量,这样不仅可以提高模型的准确性,未来在其他研究中还可以重复利用这些测序数据[91—92]。
4.3 发展基于基因功能的微生物生物地理学
在至今绝大多数的土壤微生物生物地理学研究中,分子生物学技术多被用于揭示复杂环境中微生物的系统发育多样性,而忽略了其功能多样性。实际上,真正驱动相关生态过程的是这些微生物所表现出来的功能。因此,了解自然环境中微生物功能特性的梯度(即基因地理学)将有助于从功能机理上理解微生物群落和自然地理之间的关系[93—94]。
实际上,Weiher和Keddy早在1995年就提出了基于特征的群落构建的模型,强调了物种具有的特征(而不是物种在分类学上的差异)对于群落构建的重要性[95]。这种基于功能特征的微生物生物地理学方法的潜力逐渐受到重视,因为研究功能特性与环境梯度的关系将有助于从机理上理解微生物群落和自然环境之间的关系[2,96]。随着分子生物学技术的不断发展,功能基因定量PCR技术和微阵列基因芯片技术被用于识别土壤中的功能基因,从而探索功能基因与环境梯度之间的联系[97—98]。然而,这类技术受限于已有的基因序列的知识,但自然环境中大量微生物基因的功能是未知的。宏组学(包括宏基因组、宏转录组)测序技术是实现该目标的有利工具,因为这类技术可以获得环境微生物全基因组的序列,从而实现对微生物群落的分类组成和功能特征的描述[55]。现如今,为了探索环境微生物组的功能多样性,越来越多研究者选择将宏基因组测序的手段应用于微生物生物地理研究中[99—100]。与扩增子测序相比,宏基因组测序不仅可以获得更多基因的功能信息,还能提高稀有类群的识别率[101]。需要注意的是,功能基因发生作用的前提是它们的表达,这需要结合宏转录组测序等加以研究。由此可见,结合应用各类宏组学测序方法(如Qiita平台),进行大尺度的、系统性的基因地理学分析,将是未来土壤微生物生物地理学研究的一大发展方向(图3)。更重要的是,土壤微生物生物地理学在未来还有望形成一个“基因地理学”的分支,基因地理学研究将结合宏观生态学研究方法与分子生物学手段,不仅有助于更好地理解微生物分布格局下的机制,在将来还有望对地球系统模型的构建做出重要贡献。
