全外显子组测序在9p缺失综合征诊断中的应用
2022-07-29番云华刘厚昌禹崇飞杨必清葛世军林克勤褚嘉祐杨昭庆
易 薇, 番云华, 刘厚昌, 禹崇飞, 杨必清, 葛世军, 林克勤, 褚嘉祐, 杨昭庆
(1.云南省德宏州人民医院检验科,云南 芒市 678400;2.中国医学科学院北京协和医学院医学生物学研究所医学遗传学研究室,云南 昆明 650118)
9p部分单体或9p末端缺失可导致9p缺失综合征。XY核型的男性9p缺失综合征患者在临床上会表现出男性性反转现象,导致SRXY4综合征[1]。XX核型的女性9p缺失综合征患者国内外均鲜有报道,因此9p缺失对女性子宫、卵巢等生殖系统的发生和发育造成的影响目前尚不清楚。既往对9p缺失综合征的报道多为成年男性,由于总体上报道的病例数不多,且不同病例在临床表现上呈现出异质性,除男性性反转外,目前尚未建立起较为明确的9p缺失综合征的基因型-表型关联性,其分子病理基础尚不清楚。鉴定9p缺失综合征表型相关的候选基因并探讨其表型关联性,对该病的临床诊治、预后判定和遗传咨询具有重要的意义。本研究采用染色体核型分析和全外显子组测序相结合的方法,分析1例具有子宫发育不全而无面容异常和智力语言障碍的XX核型的女性9p缺失综合征患者,以提高临床对此类疾病的认识。
1 材料和方法
1.1 研究对象
患者,女,19岁,以成年后月经未来潮就诊。患者自诉有癫痫病史。体格检查未发现身高、体质量、面容、四肢等存在异常。无语言障碍,无智力障碍,无运动发育迟缓。B超结果示幼稚子宫。实验室检查示血常规、肝肾功能、心肌酶、甲状腺功能无异常。性激素检测示性激素异常,无卵泡成熟:雌二醇20.04 pmol/L,促卵泡成熟激素2.51 IU/L,黄体生成激素1.39 IU/L,泌乳素206.9 mIU/L,孕酮0.94 nmol/L,人绒毛膜促性腺激素<0.10 IU/L。临床诊断为原发性闭经和原发性性腺功能减退,经激素治疗,闭经症状有所缓解并有持续性月经。患者的父母健康状况良好,表型正常,否认近亲婚配,否认家族遗传病史,否认孕期感染病史及放射性物质和有毒物质接触史。
1.2 方法
1.2.1 染色体G显带核型分析 经患者及其父母知情同意,采集患者及其父母外周静脉血进行淋巴细胞培养,用常规方法制备染色体,并行G显带分析。镜下计数 20个中期分裂相,并分析5个核型。染色体核型描述参照人类细胞遗传学国际命名体制2016年的标准[2]进行。
1.2.2 全外显子组测序 提取患者外周静脉血基因组DNA,经DNA片段化、末端修复、接头连接、聚合酶链反应等流程制备测序文库,在NovaSeq 6000高通量测序平台(美国Illumina公司)上进行双末端150 bp长度测序,测序深度为100×。测序结果经过滤、质控后与UCSC参考基因组(hg19,2009)进行比对,并采用美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准,结合全基因组关联研究(genome-wide association study,GWAS)数据和疾病相关的拷贝数变异(copy number variation disease,CNVD)数据库对测序结果进行分析和注释,确定突变位点对应的基因信息、功能信息及有害性等。拷贝数变异(copy number variation,CNV)结果的判定规则为:0(log2值<-1.1)、1(log2值为-1.1~-0.4)、2(log2值-0.4~0.3)、3(log2值为0.3~0.7)。其中0、1、2、3分别表示0、1、2、3个拷贝(人类中正常为2)。
2 结果
2.1 染色体核型分析结果
本例患者G显带的核型为46,XX,del(9)(p23),代表性核型见图1。其母亲的染色体核型为46,XX;父亲的染色体核型为46,XY。
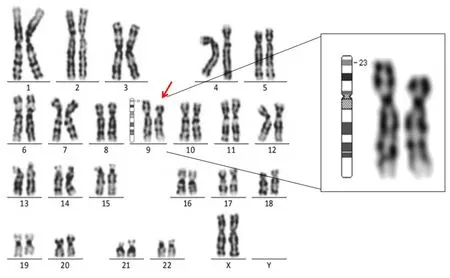
图1 本例患者的染色体核型图
2.2 全外显子组测序结果
全外显子组测序CNV分析结果显示,本例患者9p24.3-p23(24 846~13 022 661)区域存在单拷贝缺失,缺失片段长度约为13.0 Mb,该缺失区域覆盖了61个已知基因,包括DOCK8、FOXD4、KANK1、DMRT等多个与智力发育相关或性发育异常相关的基因。在全外显子组水平上未发现与临床表型相关的其他致病性CNV、碱基突变、插入和缺失。见图2。
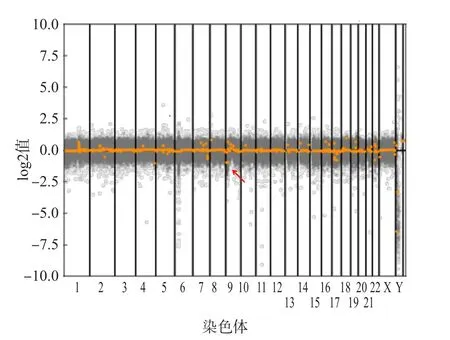
图2 患者各染色体CNV分布图
2.3 临床诊断
G显带核型分析结果提示本例患者9号染色体短臂末端缺失,经全外显子组测序分析提示该缺失区域为9p24.3-p23,缺失片段长度约为13.0 Mb,覆盖了61个已知基因。该缺失区域与9p缺失综合征的关键区域9p22.2-p23有部分重叠,属于致病性CNV。结合患者临床表现、超声及实验室检查结果,包括原发性闭经、幼稚子宫、性激素水平异常和癫痫的特征,诊断为9p缺失综合征。
3 讨论
9p缺失综合征于1973年首次被报道,典型的临床表现有三角头畸形、面部异常、智力低下、运动发育迟缓,少数患者可伴有癫痫、先天性心脏病等。男性9p缺失综合征患者可表现出外生殖器发育异常、性反转等[3]。本研究采用染色体G显带核型分析和全外显子组测序技术相结合的方法,对1例原发性闭经患者进行分析,结果显示患者染色体核型为46,XX,del(9)(p23)。采用全外显子组测序检测CNV,精确定位了染色体断裂位置和缺失基因,本例患者在9p24.3-p23(24 846~13 022 661)区域存在13.0 Mb单拷贝缺失,缺失区域涉及61个已知基因。从临床表现上看,本例女性9p末端缺失患者除原发性闭经、幼稚子宫、性激素异常、癫痫等症状外,并未表现出三角头畸形、智力语言障碍等9p缺失综合征典型的临床症状,在国内外尚未见有相似的病例报道。本研究结果为9p缺失综合征临床表型异质性提供了新的参考数据。
9p末端缺可遗传自亲代一方平衡易位的染色体,母系、父系遗传均有报道,也可能由新发染色体突变引起。本研究结果显示,患者的染色体核型为46,XX,del(9)(p23),其父母的染色体核型均未见异常,表明本例患者是新发的9p末端缺失。既往有研究发现,9p缺失综合征的关键区域为3.5 Mb长度的9p22.2-p23区域[4]。
不同9p缺失综合征患者的临床表型变异较大,可能与9p缺失的位置、片段大小或是否合并了其他染色体异常等因素有关[5]。有研究结果显示,1例男性患儿在9p24.3-p23(271 257~12 048 612)区域存在11.78 Mb单拷贝缺失,缺失区域与本例患者有较多重叠,该例患儿表现出典型的三角头畸形和智力低下[6]。ONESIMO等[7]报道了1例9pter-p24.1末端小片段缺失的男性患者,主要表现出特殊面容、癫痫、性腺发育不全等。FREDETTE等[8]报道了1例46,XY性腺发育不全患儿,其合并9p24.3-p23缺失和9p23-p21.2重复,体格检查发现有女性化外生殖器和子宫。AL ACHKAR等[9]报道了1例9p24.2-p22部分三体合并9pter-p24.2部分单体的女性患者,其表现出发育迟缓、特殊面容、原发性闭经等特征。本例患者缺失了9p24.3-p23(24 846~13 022 661)区域,与9p缺失综合征的关键区域9p22.2-p23有部分重叠,该缺失区域为致病性CNV[10]。因此,本例患者的表型也受9p缺失综合征关键缺失区域的影响,从而表现出与既往报道的9p缺失综合征相似的临床表现,包括原发性闭经、幼稚子宫、性激素水平异常和癫痫等。然而,本例患者并未表现出三角头畸形、智力语言障碍等9p缺失综合征典型的临床特征,这可能与不同病例之间缺失片段差异有关,也可能与性别因素有关。
尽管染色体显带等细胞遗传学分析为诊断9p缺失综合征提供了直观、直接的依据,但临床对缺失区域的基因型与临床表型的关联性和相关机制仍然知之甚少。本研究采用全外显子组测序技术确定了患者9p24.3-p23缺失区域覆盖了DMRT1、DMRT3、DMRT2、DOCK8、KANK1、FOXD4等多个与9p缺失综合征表型密切相关的关键基因,且在染色体其他区域并未发现其他致病性CNV、碱基插入和缺失。9p末端9p24.3上的DMRT(DMRTl、DMRT2、DMRT3)是维持男性睾丸分化所必需的基因。男性9p末端缺失可导致DMRT1基因单倍剂量不足,引起男性性反转[11]。DMRT基因在女性泌尿生殖系统发育中的作用目前尚不明确。有研究结果显示,DMRT1基因在人类女性胚胎的卵母细胞中有表达,其缺失可能与女性卵巢早衰有关,可引起育龄女性原发性不孕[12-13]。因此,9p24.3-p23缺失导致DMRT1基因单倍剂量不足可能是导致本例患者出现子宫发育不良、原发性闭经、性激素异常的主要原因。同时,DOCK8、FOXD4、KANK1等基因与智力发育相关,DOCK8基因缺失可引起智力低下和癫痫[7],FOXD4基因缺失可能导致智力低下或语言障碍,而KANK1基因缺失可导致家族性脑瘫[14]。因此,DOCK8基因缺失可能是本例患者表现出癫痫的主要原因,但本例患者并未观察到智力障碍,原因可能是不同患者缺失区域所涉及的DOCK8等智力发育相关基因及其附近多个基因的片段大小不同,对DOCK8基因的表达产生不同的综合作用,从而导致不同的9p缺失综合征患者在智力发育方面表现出较大的差异。此外,有研究结果表明,位于9p22.3区域的CERl基因是9p缺失综合征患者三角头畸形的关键基因[15]。本例患者9p24.3-p23缺失片段并未覆盖CERl基因,这可能是本例患者未表现出三角头畸形的原因。本研究结果为揭示CER1基因型和表型的关联性提供了更多的参考依据。
综上所述,9p末端缺失导致DMRT1基因单倍剂量不足可能是导致本例患者幼稚子宫、原发性闭经的主要致病因素。本研究为原发性闭经、性发育异常和染色体病的临床诊断提供了更多线索,进一步揭示了9p缺失综合征的临床病理特征。由于9p缺失综合征较为罕见且具有表型异质性,通过更多病例的收集和分析将有助于明确DMRT1等基因在女性子宫及其他泌尿生殖系统发育中的作用和机制,为该病的诊疗和遗传咨询提供更多的参考依据。
