伊曲康唑胶囊有关物质测定方法优化*
2022-07-27张秉华杜亚俊衷红梅牛龙青赵蒲中
张秉华,杜亚俊,衷红梅,牛龙青,赵蒲中
(陕西省食品药品检验研究院,陕西 西安 710065)
伊曲康唑为三唑类抗真菌药物,主要通过干扰真菌细胞膜麦角固醇的生物合成来抑制真菌增殖、促进真菌死亡,具有抗菌谱广、毒性低的特点,临床适用于浅表真菌感染和深部真菌引起的系统感染,及其他抗真菌药物不适用或无效的系统性感染[1−4]。伊曲康唑胶囊为我院2021年国家评价性抽验品种,抽验执行2015年版《中国药典(第一增补本)》[5]和2020 年版《中国药典(二部)》[6]中有关物质检查方法。在实际检验中发现,系统适用性溶液制备困难,难以达到系统适用性要求;部分样品相邻杂质峰分离效果欠佳,影响结果。查阅文献[7]发现,药典中有关物质检测方法为2010 年前起草,杂质E峰的相对保留时间可能存在偏差,企业反映该相对保留时间的杂质峰无法通过强制降解试验得到,造成在未达到系统适用性要求的情况下开展检验。此外,随着伊曲康唑生产工艺的变化和对其杂质谱研究的深入,药典中相关物质检测方法仅对单个杂质和杂质总量进行控制,难以真实反映各企业的产品质量。本研究中采用高效液相色谱法对现行有关物质检测方法进行改进,以期建立灵敏度高、分离效果好、检测效率高、对环境友好的测定方法。现报道如下。
1 仪器与试药
1.1 仪器
Agilent 1260型高效液相色谱仪(美国Agilent公司)、Shimadzu LC −2030 3D 型高效液相色谱仪(日本Shimadzu 公司);XPE205 型、ME204 型电子天平(瑞士Mettler Toledo 公司);Milli −Q 高纯水机(美国Millipore公司)。
1.2 试药
伊曲康唑对照品(批号100631 −201402,含量99.2%)、杂质A 对照品(批号130686 −201501,含量99.5%)、杂质B 对照品(批号130687 −201501,含量97.5%)、杂质C 对照品(批号130688 −201501,含量96.3%)、杂质D 对照品(批号130689 −201501,含量95.1%)、杂质E 对照品(批号130690 −201501,含量97.1%)、杂质F 对照品(批号130691 −201501,含量98.0%)、杂质G 对照品(批号130692 −201501,含量95.5%),均购自中国食品药品检定研究院;伊曲康唑杂质H 对 照 品(企 业D,批 号WVLA_0062_003_1,含 量97.2%)。伊曲康唑胶囊为2021年国家评价性抽验样品,此次抽样涉及A,B,C,D 4个生产企业,各3批。原辅料为生产企业A,B,C提供;四氢呋喃、甲醇、乙腈均为色谱纯,四丁基硫酸氢铵、乙酸铵均为分析纯,水为高纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Aglient Proshell 120 EC −C18柱(150 mm ×4.6 mm,2.7 μm);流动相:0.01 mol/L乙酸铵溶液(A)−乙腈(B),梯度洗脱(0~3 min 时62%A,3~6 min 时62%A→45%A,6~11 min 时45%A;11~19 min 时45%A→34%A,19~36 min 时34%A;36~38 min 时34%A→62%A,38~40 min时62%A);流速:0.8 mL/min;检测波长:260 nm;柱温:30 ℃;进样量:10 μL。
2.2 溶液制备
稀释剂:稀释剂Ⅰ为甲醇−乙腈(3∶1,V/V);精密量取稀释剂Ⅰ700 mL,加0.01 mol / L 乙酸铵溶液300 mL,混匀,即得稀释剂Ⅱ。
对照品溶液:取杂质B 对照品0.012 79 g、杂质F 对照品0.0129 2 g、杂质G 对照品0.011 98 g,精密称定,置100 mL 容量瓶中,加稀释剂Ⅰ70 mL 使溶解,加稀释剂Ⅱ定容,摇匀,即得杂质B,F,G 的混合对照品溶液。取杂质H对照品0.004 30 g,精密称定,置20 mL容量瓶中,加稀释剂Ⅰ14 mL 使溶解,加稀释剂Ⅱ定容,摇匀,即得杂质H 对照品溶液。分别取杂质A 对照品、杂质B对照品、杂质C 对照品、杂质D 对照品、杂质E 对照品、杂质F对照品、杂质G对照品、杂质H对照品约12 mg,精密称定,置100 mL容量瓶中,加稀释剂Ⅰ70 mL使溶解,再加稀释剂Ⅱ定容,摇匀;精密量取1 mL,加入伊曲康唑对照品40 mg,置100 mL容量瓶中,加稀释剂Ⅰ70 mL使溶解,再加稀释剂Ⅱ定容,摇匀,制成每1 mL中含各杂质1.2 μg和含伊曲康唑0.4 mg的混合对照品溶液。
供试品溶液:取伊曲康唑胶囊内容物适量(约相当于伊曲康唑100 mg),精密称定,置250 mL容量瓶中,加稀释剂Ⅰ175 mL 使溶解,加稀释剂Ⅱ定容,摇匀,滤过,取续滤液,即得。
对照溶液:精密量取供试品溶液1 mL,置25 mL容量瓶中,加稀释剂Ⅰ17.5 mL,加稀释剂Ⅱ定容,摇匀;精密量取1 mL,置20 mL容量瓶中,加稀释剂Ⅰ14 mL,加稀释剂Ⅱ定容,摇匀,即得。
阴性对照品溶液:按辅料用量最多的生产企业的处方工艺及比例制备缺伊曲康唑的阴性样品,按供试品溶液制备方法制得阴性对照品溶液。
2.3 方法学考察
专属性试验:分别取2.2项下混合对照品溶液和阴性对照品溶液适量,按2.1 项下色谱条件进样测定,记录色谱图。可见,杂质C与杂质D为异构体,保留时间相同,其余各峰之间分离度均大于1.5,且阴性对照无干扰。详见图1。
线性关系考察:分别取伊曲康唑、杂质B 对照品、杂质C 对照品、杂质D 对照品、杂质F 对照品、杂质G 对照品、杂质H 对照品适量,精密称定,加入70%体积的稀释剂Ⅰ使溶解,再加入稀释剂Ⅱ定容,倍比稀释,制成质量浓度分别为0.3,0.6,1.2,1.8,2.4 μg/mL 的系列混合对照品溶液,按2.1 项下色谱条件进样测定,记录峰面积。以质量浓度(X,μg/ mL)为横坐标、峰面积(Y)为纵坐标进行线性回归,得回归方程,以斜率计算相对校正因子。结果见表1。
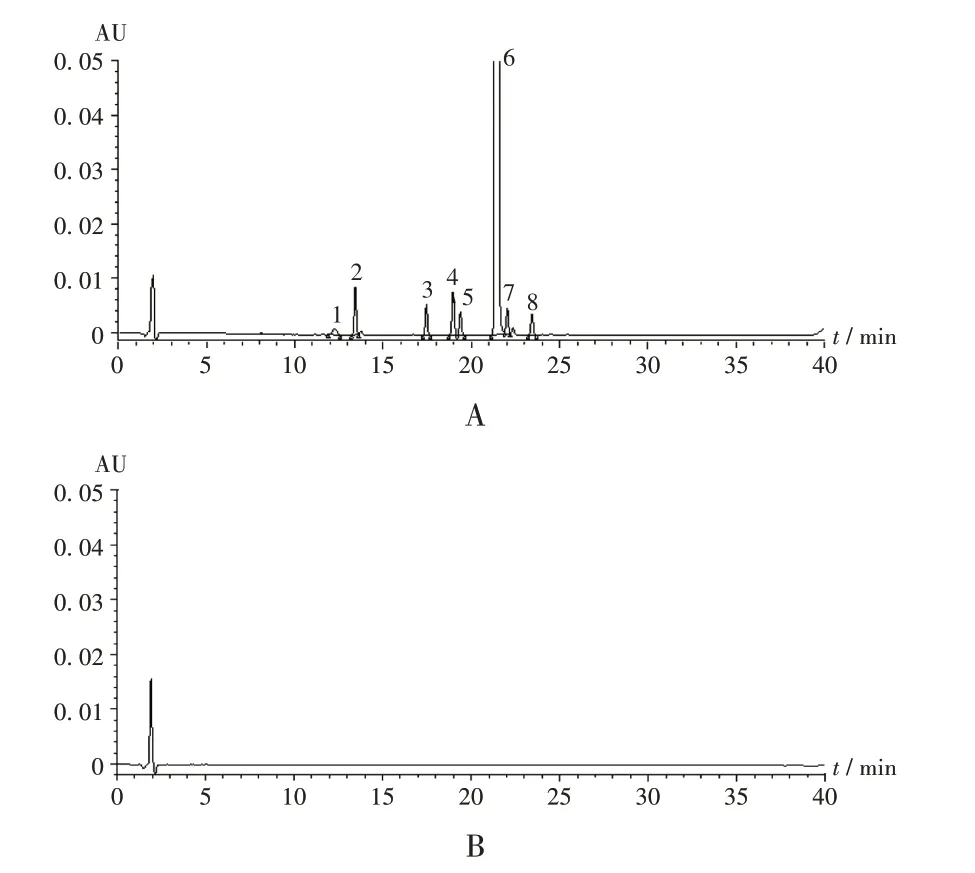
1.杂质H 2.杂质A 3.杂质B 4.杂质C(或杂质D) 5.杂质E 6.伊曲康唑 7.杂质F 8.杂质GA.混合对照品溶液 B.阴性对照品溶液图1 高效液相色谱图1.Impurity H 2.Impurity A 3.Impurity B 4.Impurity C/D 5.Impurity E 6.Itraconazole 7.Impurity F 8.Impurity GA.Mixed reference solution B.Negative reference solutionFig.1 HPLC chromatograms
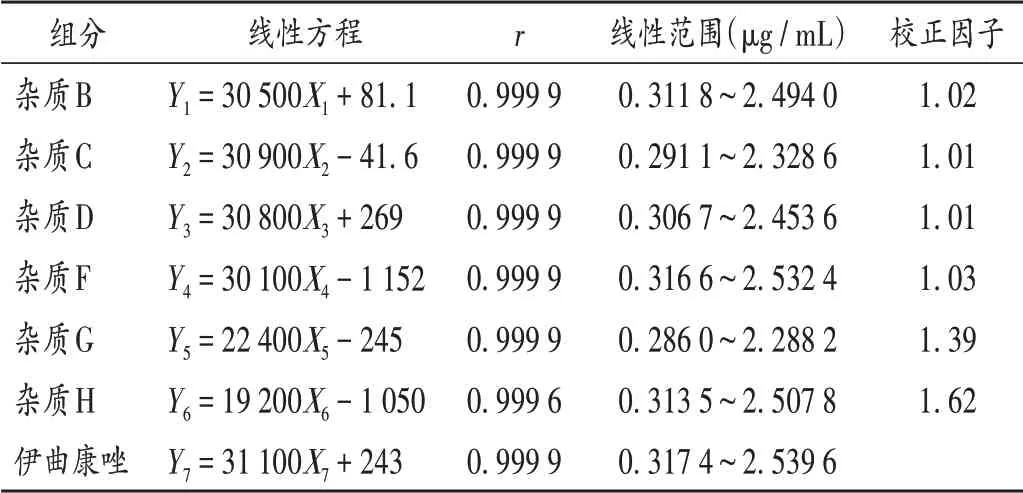
表1 线性关系考察结果(n=5)Tab.1 Results of the linear relation test(n=5)
定量限与检测限考察:取线性关系考察项下的系列混合对照品溶液,倍比稀释,按2.1 项下色谱条件进样测定,以信噪比为3∶1 和10∶1 时的质量浓度分别作为检测限和定量限。结果伊曲康唑、杂质B、杂质C、杂质D、杂质F、杂质G、杂质H 的检测限分别为0.38,0.37,0.35,0.37,0.38,0.57,1.25 ng,定量限分别为1.27,1.25,1.16,1.23,1.27,1.37,3.76 ng。
重复性试验:取企业D 同一批样品适量,共6 份,按2.2 项下方法制备供试品溶液,按2.1 项下色谱条件进样测定,记录峰面积并计算含量。由于样品中单个杂质含量较低(均小于0.05%),6 份样品中单个杂质含量测定结果的RSD均小于3.85%,杂质总量的RSD为0,表明方法重复性良好。
加样回收试验:参考欧洲药典和美国药典相关标准及文献[8],以0.3%为各杂质限度,制备样品中检出率较高的杂质B、杂质F、杂质G、杂质H 的回收率溶液。称取各杂质含量已知的同一批样品适量(约相当于伊曲康唑100 mg),精密称定,置250 mL容量瓶中,分别加入杂质B,F,G 的混合对照品溶液和杂质H 对照品溶液适量,制成杂质含量分别为0.12%,0.3%,0.5%的溶液,按2.1项下色谱条件进样测定,记录峰面积,并计算加样回收率[9]。结果见表2。

表2 加样回收试验结果(n=9)Tab.2 Results of the recovery test(n=9)
2.4 样品检测
取4个企业的12批样品适量,按2.2项下方法制备供试品溶液,分别加入对照溶液,按2.1 项下色谱条件进样测定,记录峰面积,并采用主成分自身对照法计算杂质B,C,D,F 和其他单个杂质含量,杂质G 和杂质H含量按加校正因子的主成分自身对照法计算,含量低于0.05%的杂质忽略不计。结果见表3。
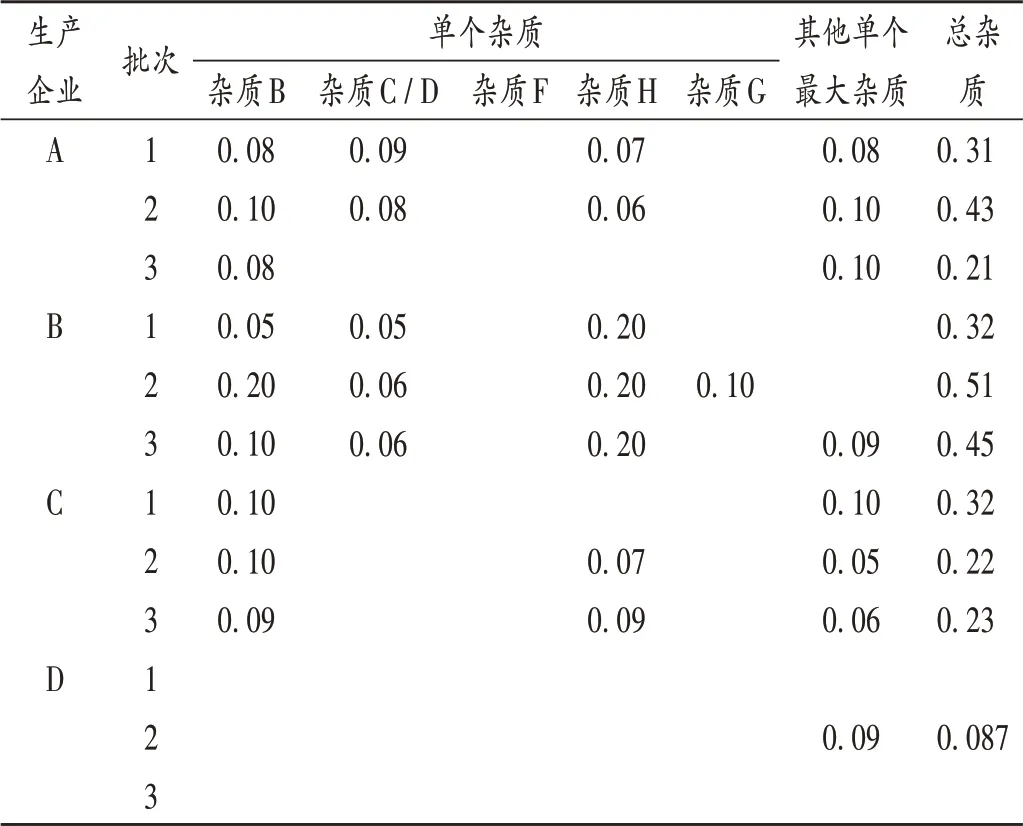
表3 伊曲康唑胶囊有关杂质检测结果(%)Tab.4 Results of content determination of related substance in Itraconazole Capsules(%)
2.5 不同测定方法结果比较
采用药典[5−6]中有关物质检查方法,对4 个企业同批号的12 批样品进行含量测定,并与本研究中新建方法结果进行比较。结果表明,2 种方法检测的企业B,C,D 样品含量的结果基本一致,采用新建方法测得企业A 3 批样品的杂质总量(0.31%,0.43%,0.21%)高于药典中的方法(0.25%,0.28%,0.15%)。比较企业A 同批号样品色谱图(图2)发现,新建方法的杂质检出个数更多。
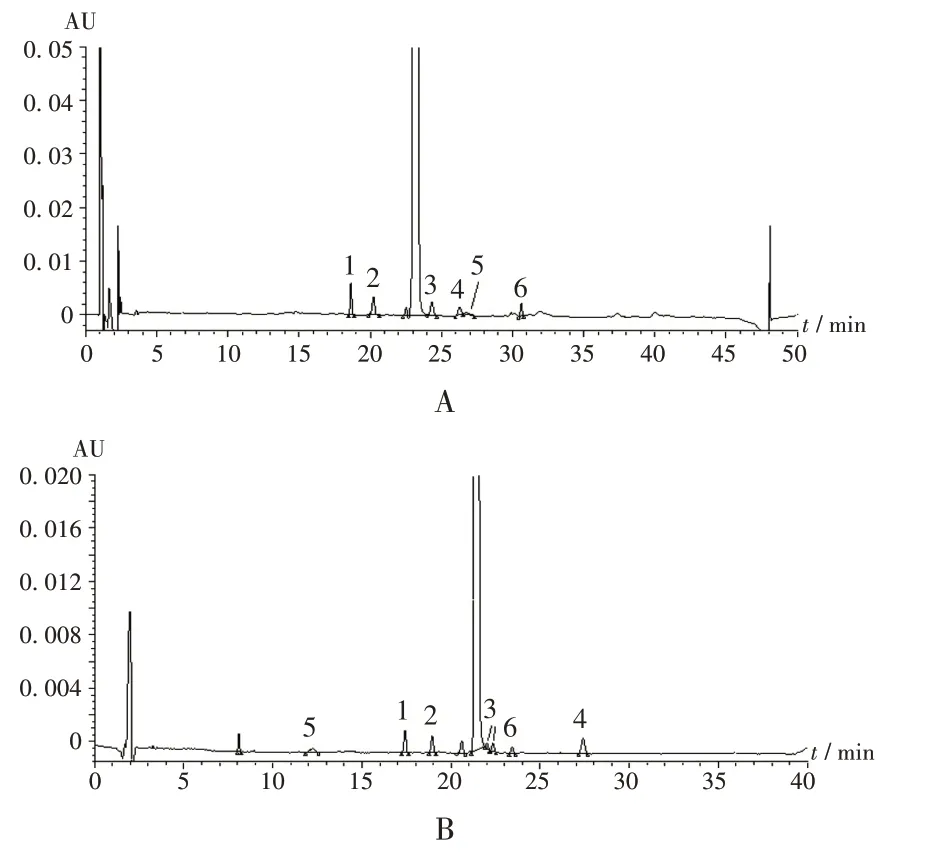
1.杂质B 2.质杂C和D 3.杂质F与未知杂质 4.未知杂质5.杂质H 6.杂质GA.现行标准方法(主药质量浓度2 mg/mL) B.新建方法(主药质量浓度0.4 mg/mL)图2 不同方法测定同批号伊曲康唑胶囊有关物质色谱图1.Impurity B 2.Impurity C/D 3.Impurity F and unknown impurity 4.Unknown impurity 5.Impurity H 6.Impurity GA.Current standard method(the mass concentration of main drug is 2 mg/mL) B.New method(the mass concentration of main drug is 0.4 mg/mL)Fig.2 Chromatograms of related substances in Itraconazole Capsules with same batch number determined by different methods
3 讨论
3.1 检测波长选择
预试验中对混合对照品溶液和供试品溶液进行全波长扫描,发现伊曲康唑、杂质A~G 在(260 ± 2)nm波长处、杂质H 在(248 ± 2)nm 波长处有最大吸收峰,为保证多数杂质的检出灵敏度,选择260 nm 作为检测波长。
3.2 耐用性考察
取混合对照品溶液和供试品溶液,通过更换不同型号高效液相色谱仪(Agilent 1260 型、岛津LC2030C 3D 型),不同色谱柱[Aglient Proshell 120 EC −C18柱(150 mm × 4.6 mm,2.7 μm)、Welch Boltimate EXT −C18柱(150 mm×4.6 mm,2.7 μm)],以及改变柱温(±3 ℃),各杂质均可有效分离,表明方法耐用性良好。
3.3 出峰情况
药典中有关物质检测采用甲酸破坏法制备系统适用性溶液,检验中发现主峰前后的杂质峰难以强制降解得到,生产企业均反映受此问题困扰。本研究中尝试多种方法制备系统适用性溶液效果均不佳,建议采用杂质C(或杂质D)、杂质E、伊曲康唑、杂质F 制备系统适用性溶液,杂质C(或杂质D)峰与杂质E峰、伊曲康唑峰与杂质F峰分离度应符合要求。
综上所述,本研究前期对4 个生产企业131 批样品的杂质进行含量测定,结果杂质B、杂质F、杂质G、杂质H 的检出率较高,故本研究中主要对这4 种杂质的含量测定进行方法学考察。结果显示,所建有关物质检测方法灵敏度高、分离效果好,运行时间短,流速低,有机溶剂用量少,对环境更友好,可满足4 个生产企业的伊曲康唑胶囊产品质量控制要求。
