超高效液相色谱-串联质谱测定婴幼儿纺织用品中的喹啉和异喹啉残留量
2022-07-23李志刚蒋少军
李志刚, 蒋少军, 戴 鸽
(1.浙江工业职业技术学院鉴湖学院,浙江绍兴 312000;2.宁波申洲针织有限公司,浙江宁波 315000)
喹啉又名苯并吡啶,无色液体,属芳香类化合物。异喹啉是喹啉的同分异构体,喹啉和异喹啉都是重要的生物碱,具有较强的生理活性,被广泛用于橡胶、印染、精细化工等领域。喹啉经过硝化、还原得到硝基喹啉,可用作皮毛染色剂及纺织品染色辅助中[1]。喹啉和异喹啉均属中等毒性,其蒸气对喉、鼻有刺激性,吸入后可出现恶心、头晕等症状,对呼吸系统和皮肤有刺激性,对眼睛有严重伤害,对生物体有致癌、致畸、致突变风险,被公认为是有毒难降解的有机化合物。欧洲化学品管理局(ECHA)已将喹啉归类为致癌、致突变或致生殖毒性物质。最新发布的STANDARD 100中明确规定它们的限量值为50 mg/kg[2 - 5]。因此,对纺织品尤其是婴幼儿纺织品中喹啉和异喹啉残留量进行监测非常有必要。目前国内未见有相关的标准检测方法。
目前,喹啉和异喹啉常用的检测方法有高效液相色谱法[2,5]、气相色谱法[6]、气相色谱-质谱法[1,3,4,7]以及液相色谱-质谱联用法[8]。色谱法以其设备价格低,通用性好等优点,广泛应用于食品、水质、化妆品、纺织品等领域的检测,但是由于液相色谱和气相色谱法都是以化合物的保留时间作为定性依据,在复杂基质中容易受到共流出化合物的干扰,影响测定结果的准确性。液相色谱-串联质谱法可以通过对母离子扫描、子离子扫描和中性丢失扫描,有效排除基质离子产生的非目标碎片离子的干扰,优化质谱检测信号,具有高的选择性和灵敏度,是近些年来发展十分迅速的分析技术[9 - 11]。本实验采用甲醇对样品进行提取,以分散固相萃取[12 - 15]对提取液进行净化,结合液相色谱-串联质谱法测定婴幼儿纺织用品中喹啉和异喹啉的残留量,取得良好的实验效果。
1 实验部分
1.1 实验仪器
Prominence UFLC XR型高效液相色谱仪(日本,岛津株式会社);AB 5500型三重四极杆质谱仪(美国,AB SCIEX公司)以及数据分析系统;HGC-24型氮吹仪(天津恒奥科技有限公司);1-14K离心机(德国,SIGMA公司);Milli-Q纯水系统(美国,Millipore公司);全玻璃溶剂过滤器(美国,Waters公司);DIGITALVORTEX-涡旋混合器(美国,SI公司);XS205电子分析天平(瑞士,梅特勒公司)。
1.2 试剂和材料
喹啉(CAS:91-22-5,纯度98.5%)、异喹啉(CAS:119-65-3,纯度96.3%)(北京曼哈格生物科技有限公司);乙腈、甲醇、甲酸(色谱纯,赛默飞世尔科技有限公司);氨水(色谱纯,上海阿拉丁生化科技股份有限公司)。SCX吸附剂(粒径60 nm,上海麦克林生化科技有限公司);滤头(0.22 μm,上海安谱实验科技股份有限公司);一次性注射器(常州悦康医疗器材有限公司);50 mL聚丙烯离心管(浙江拱东医疗科技有限公司)。
1.3 标准储备液的配制
分别准确称取10.00 mg喹啉和异喹啉标准物质于2个10 mL容量瓶中,用甲醇溶解、稀释并定容,配制成1.0 mg/mL的标准储备溶液。准确吸取喹啉和异喹啉标准储备溶液(1.0 mg/mL)各10.0 μL于10 mL容量瓶中,用甲醇定容至刻度,配制成1.0 mg/L的混合标准应用液。
1.4 色谱-质谱条件
色谱条件:Waters ACQUITY UPLC BEH C18柱(150 mm×2.1 mm,1.7 μm);流动相:0.1%甲酸的水溶液(A)和0.1%甲酸的乙腈溶液(B),A/B=95/5,等度洗脱;流速:0.35 mL/min;柱温:40 ℃;进样体积:5 μL。
质谱条件:电喷雾离子源(ESI);正离子扫描;多反应监测模式(MRM)检测;电喷雾电压(IS):4 500 V;雾化气压力(GS1):50.0 psi;辅助气流速(GS2):50.0 psi;气帘气压力(CUR):20.0 psi;碰撞气(CAD):8.0 psi;离子源温度(TEM):500 ℃;扫描时间:50 ms;碰撞室出口电压(CXP):11.0 V;碰撞室入口电压(EP):10.0 V。定性离子对、定量离子对、碰撞气能量(CE)及去簇电压(DP)见表1。喹啉和异喹啉标准溶液的MRM图见图1。
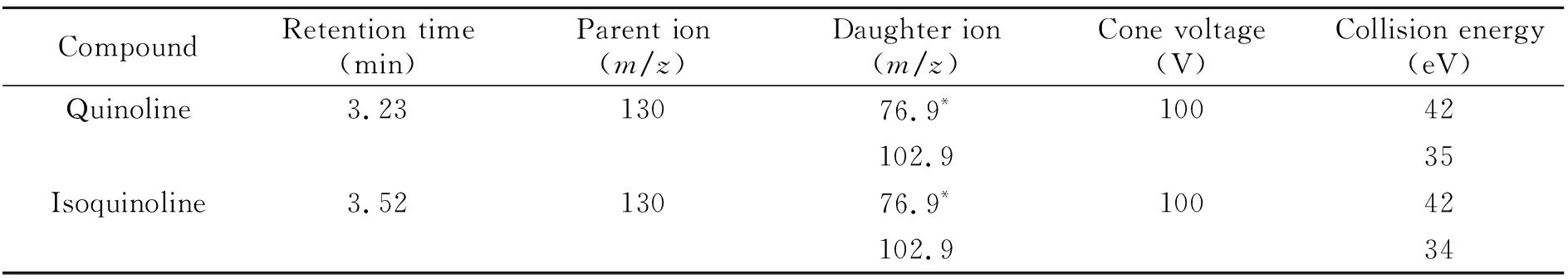
表1 质谱分析参数
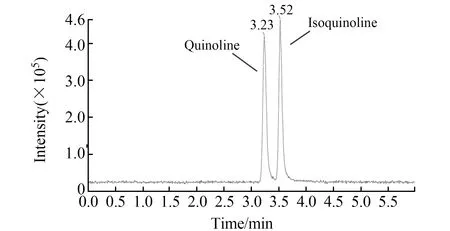
图1 喹啉和异喹啉标准溶液的MRM色谱图Fig.1 MRM of chromatogram of quinoline and isoquinoline
1.5 样品处理
准确称取1.0 g(精确至0.1 g)剪碎、混匀的样品,装入50 mL离心管中,加入20 mL甲醇,立即密闭,在40 ℃水浴超声提取30 min,取出,冷却至室温,8 000 r/min,离心5 min。移出上清液于另一个50 mL离心管中,在提取液中加入25 mg SCX吸附剂,混漩1 min后,8 000 r/min离心5 min,小心移去上清液,在SCX吸附剂中加入0.5%氨水甲醇1 mL,混漩1 min后,8 000 r/min离心5 min,小心吸出上清液,在SCX吸附剂中再次加入0.5%氨水甲醇1 mL,同样操作一次。合并上清液,氮吹至近干,用1 mL流动相复溶后,过0.22 μm滤膜,进样测定。
2 结果与讨论
2.1 流动相的选择
实验首先选择水-甲醇和水-乙腈作为流动相体系,考察目标化合物的色谱/质谱行为。结果发现,喹啉在这两种流动相条件下,其峰形对称,响应相当。考虑到目标化合物在正离子模式下具有较强的质谱响应,实验在流动相中添加适量甲酸,提供H+,以提高灵敏度。实验结果证实,在流动相中添加甲酸可以提高待测物的灵敏度。实验比较了添加0.1%和0.2%甲酸对灵敏度的影响,结果显示两者灵敏度相当,故选择水-乙腈(均含0.1%甲酸)作为色谱流动相。本实验的两个目标化合物是同分异构体,质谱裂解规律一致,只能靠液相分离来进行定性,通过对色谱柱、流动相比例以及流速的选择和优化,最终确定的液相条件见“1.4”。
2.2 质谱条件优化
为获得喹啉和异喹啉的最优质谱参数,实验用针泵直接进样,分别测定了喹啉和异喹啉标准溶液在ESI+和ESI-模式下的质谱信号强度。结果显示,化合物在ESI+模式下的质谱信号强于ESI-模式下的质谱信号,可能是待测化合物结构中均含有N原子所致,使其容易得到质子而形成[M+H]+准分子离子峰。随后,分别对各待测物母离子进行碰撞诱导电离,获得二次裂解碎片离子。对各化合物碎片离子进行碰撞能量的优化,选择丰度高、干扰小的两对离子进行定性定量分析。喹啉和异喹啉在正离子模式下均形成[M+H]+的分子离子峰m/z130.0,通过调节合适的去簇电压和碰撞能量,主要产生m/z76.9和m/z102.9的碎片离子,图2为喹啉和异喹啉的二级质谱图。优化后的各化合物的质谱参数见表1。
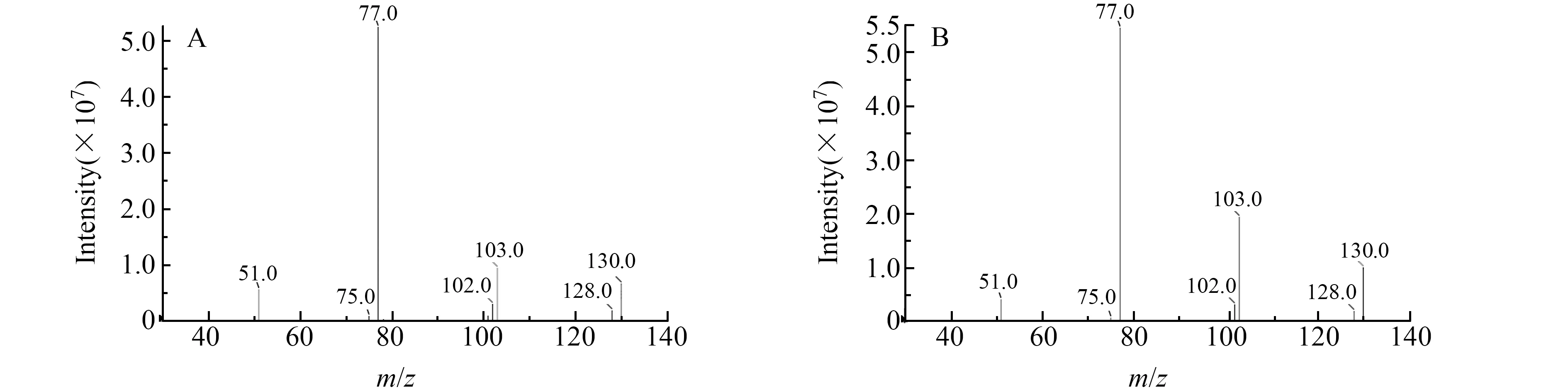
图2 喹啉和异喹啉的二级质谱图(A)喹啉(B)异喹啉Fig.2 MS/MS of quinoline and isoquinoline(A) Quinoline(B)Isoquinoline
2.3 分散固相萃取条件优化
2.3.1 分散吸附剂选择本实验选择常用的含羧酸基团的WCX、含苯磺酸基团的SCX、含丙磺酸基团的PRS、含二醇基基团的Diol以及含二氨基基团的PSA分散吸附剂对待测化合物的吸附能力进行试验。实验结果显示,含羧酸基团的WCX其平均吸附效率为40%,含苯磺酸基团的SCX和含丙磺酸基团的PRS其平均吸附效率均为99%,Diol和PSA几乎不吸附目标化合物,结果见图3。实验选择SCX和PRS再进行优化。
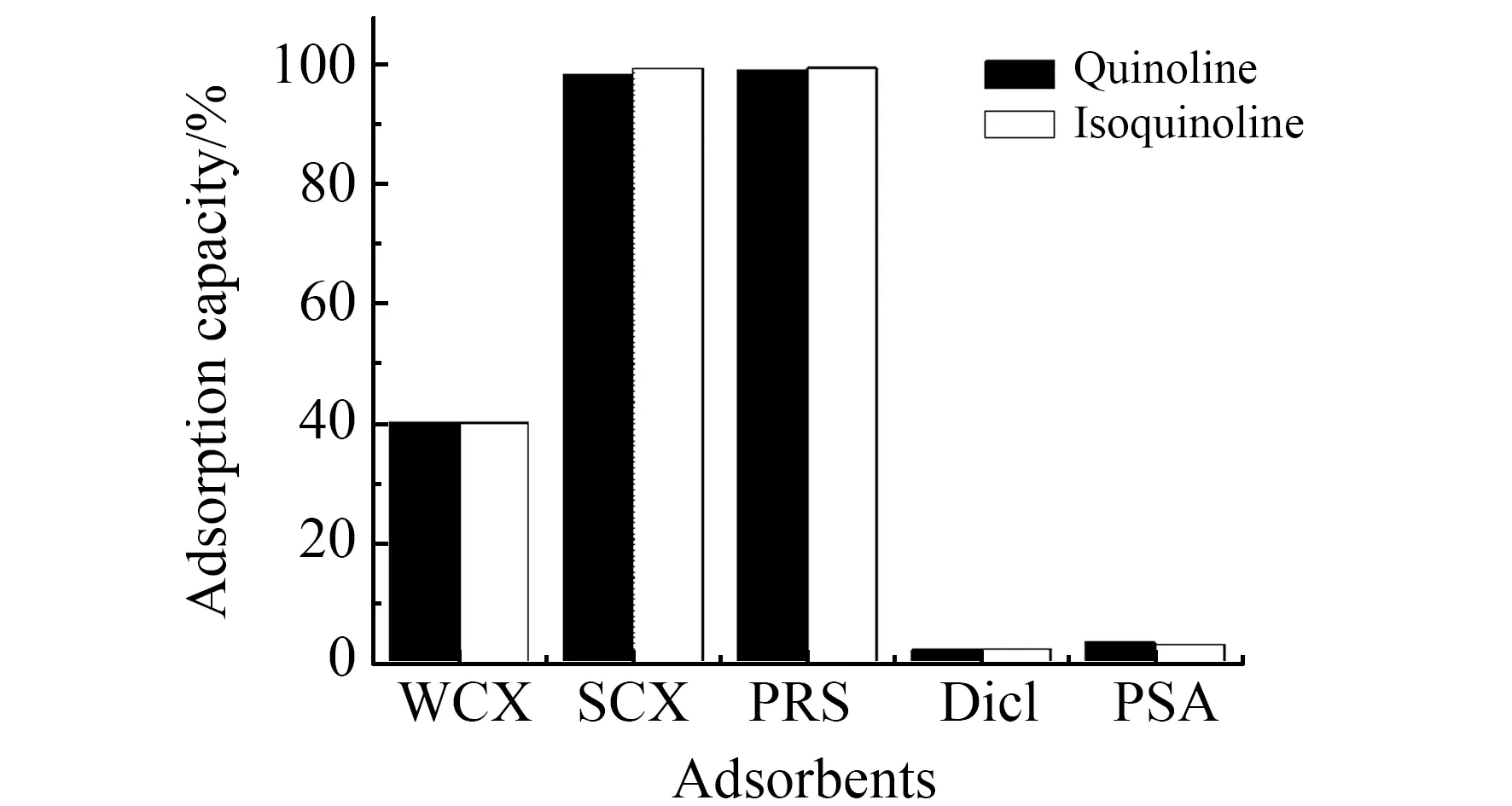
图3 不同吸附剂对目标化合物的吸附能力Fig.3 Adsorption capacity of different adsorbents on target compounds
2.3.2 吸附剂用量的选择在12份20 mL喹啉和异喹啉混合标准溶液(0.5 mg/L)中,分别加入不同质量的SCX或PRS吸附剂,混旋1 min后,8 000 r/min离心5 min,取上清液测定。两种吸附剂的吸附效率如图4所示,SCX的吸附能力略强于PRS,实验选择SCX作为吸附剂,添加量为25 mg。
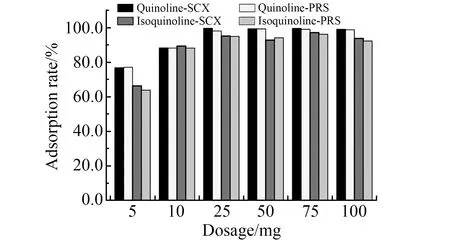
图4 吸附剂用量优化Fig.4 Optimization of adsorbent dosage
2.3.3 吸附时间选择在5份20 mL喹啉和异喹啉混合标准溶液(0.5 mg/L)中,各加入25 mg的SCX吸附剂,设置不同的混旋时间(1 min、2 min、5 min、8 min、15 min),混旋后,8 000 r/min离心5 min,取上清液测定,考察其吸附效果。实验结果显示,混旋1 min就可以吸附溶液中的待测化合物。
2.3.4 解析液中氨水的比例选择取吸附了喹啉和异喹啉之后的SCX吸附剂6份,分别加入1 mL含不同氨水比例的甲醇洗脱液,考察其洗脱效果。如图5所示,0.5%氨水甲醇就能够完全解析吸附剂中的目标化合物。实验选择洗脱溶液为0.5%氨水甲醇。
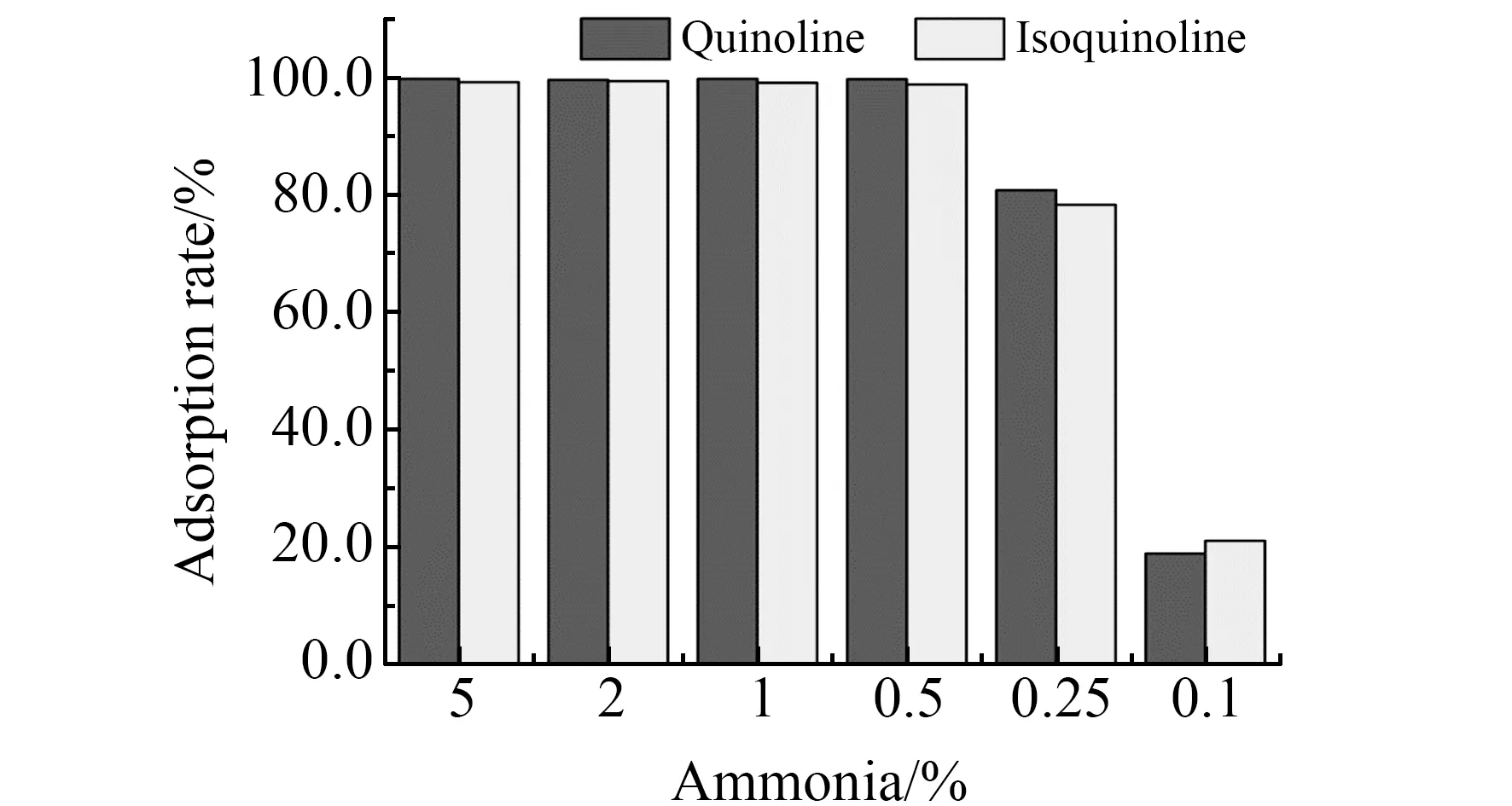
图5 不同氨水比例的洗脱能力Fig.5 Elution ability of different ammonia content
2.3.5 解析时混旋的时间选择取吸附了喹啉和异喹啉之后的SCX吸附剂5份,分别加入0.5%氨水甲醇1 mL,随后各自混旋1 min、2 min、5 min、8 min以及15 min,考察其洗脱效果。实验结果显示,1 min就能够使目标化合物完全解析出来,实验选择解析时间为1 min。
2.4 基质效应
基质效应的评价一般采用相同浓度的基质标准溶液与溶剂标准溶液的比值进行评价。比值越接近1,说明基质效应越小。为考察基质效应,实验分别取棉麻、全棉和丝绸为面料的婴幼儿服装的空白混匀样品各6份,按“1.5”进行样品处理后,再加入适量的喹啉和异喹啉的混合标准应用液(1.0 mg/L),配制成含喹啉和异喹啉均为1.0 μg/L、5.0 μg/L和50.0 μg/L的基质加标溶液,按“1.4”进行测定,记录峰面积为B。再用流动相为溶剂,配制成含喹啉和异喹啉均为1.0 μg/L、5.0 μg/L和50.0 μg/L的质控(QC)溶液,按“1.4”进行测定,记录峰面积为A,基质效应=B/A×100%。基质效应结果见表2,喹啉和异喹啉基质效应为93.8%~98.1%。可见待测化合物在样品中存在非常微弱的基质抑制,可以忽略,本实验采用标准曲线对目标化合物进行定量。
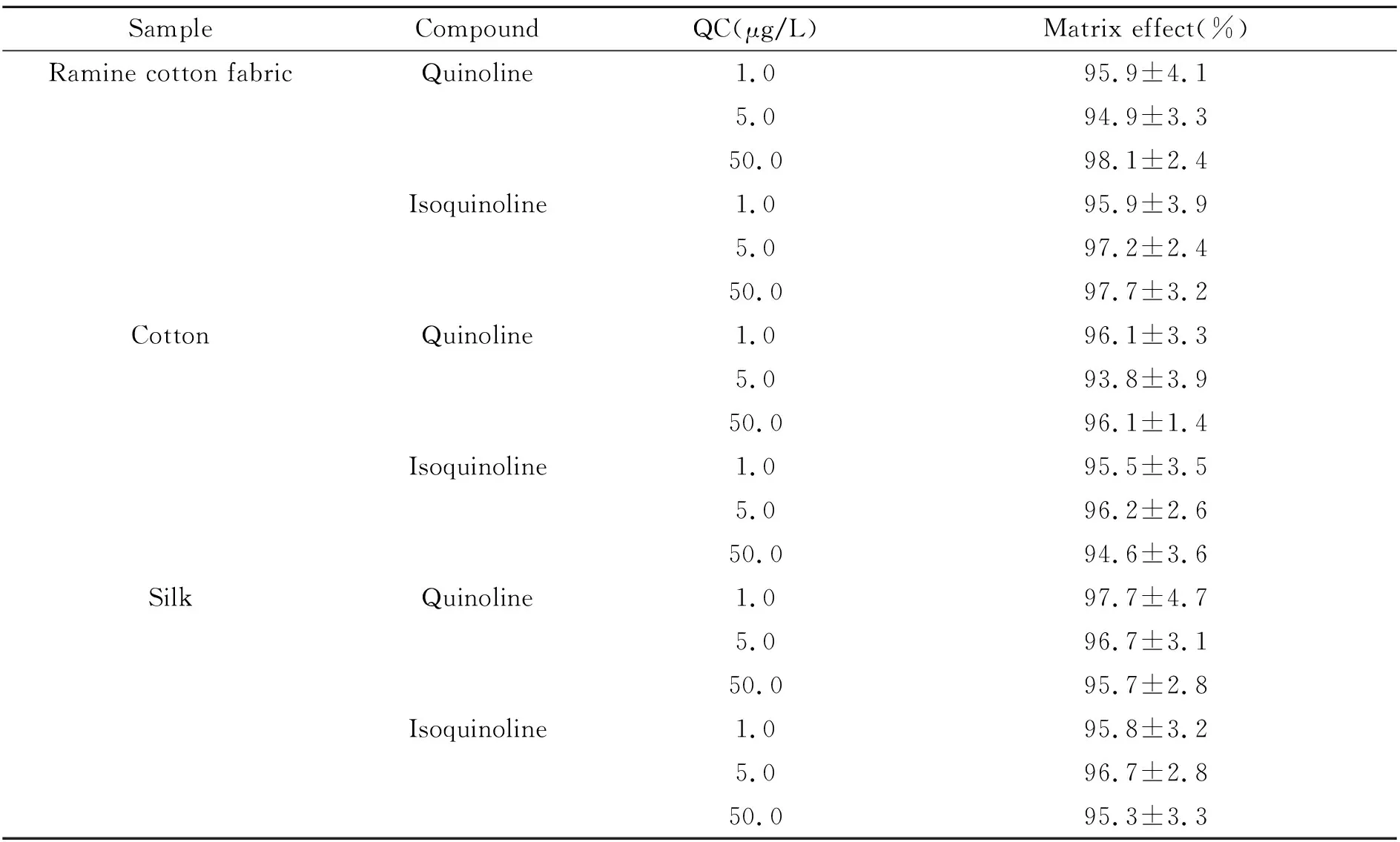
表2 基质效应(n=6)
2.5 方法学评价
2.5.1 线性范围和检出限取适量“1.3”配制的混合标准应用液(1.0 mg/L),用流动相稀释制备得到混合标准溶液系列,其浓度为0.5 μg/L、1.0 μg/L、5.0 μg/L、10.0 μg/L、20.0 μg/L、100.0 μg/L,进样测定,以待测物的峰面积(y)与对应的质量浓度(c,μg/L)进行线性回归,得到回归方程和相关系数见表3。实验结果表明,喹啉和异喹啉均在0.5~100.0 μg/L浓度范围内呈良好的线性关系。
取混合均匀的阴性样品(棉麻、全棉和丝绸)各1.0 g于50 mL离心管中,分别加入喹啉和异喹啉的混合标准应用液(1.0 mg/L)各1.0 μL,静置0.5 h后按“1.5”样品测定操作,依据MRM色谱峰的信噪比(S/N)等于3倍确定检出限(LOD),S/N等于10倍确定定量限(LOQ),得到喹啉和异喹啉的LOD和LOQ分别为0.16 μg/kg和0.5 μg/kg(表3)。

表3 化合物的线性相关系数、线性范围、检出限(LOD)和定量限(LOQ)
2.5.2 回收率和精密度分别取棉麻、全棉和丝绸空白样品各9份,样品混匀之后,各称1.0 g于50 mL离心管中,再准确加入一定量的混合标准应用液(1.0 mg/L),制备成高、中、低三个浓度水平的质量控制样品各3份,静置0.5 h后按“1.5”样品测定操作。每份样品重复测定6次,计算方法的准确度(以平均回收率计)和相对标准偏差(RSD),结果见表4。喹啉和异喹啉在样品中的回收率分别为90.2%~104.5%和91.6%~105.6%,RSD分别为2.4%~5.3%和3.8%~6.3%。
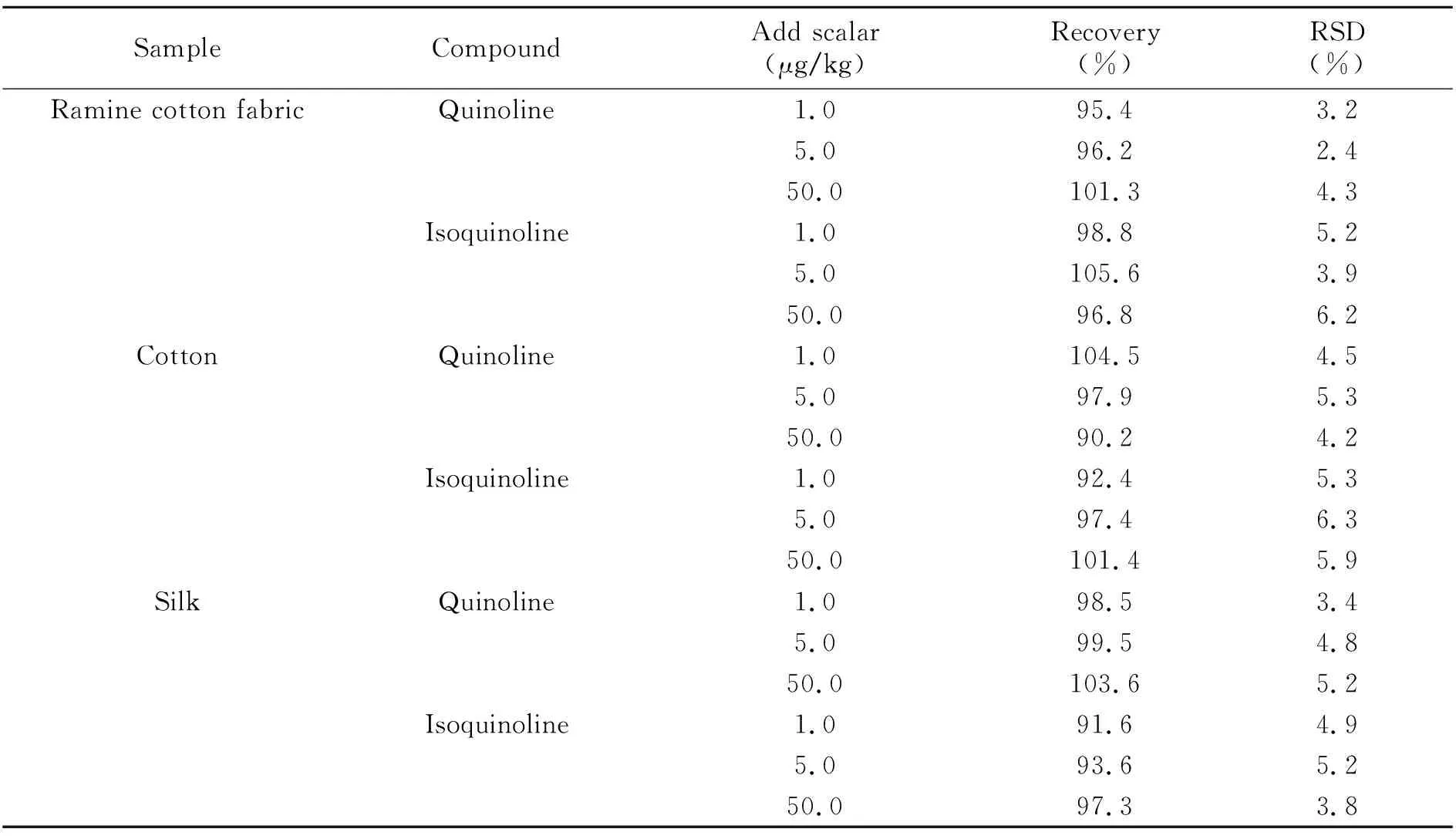
表4 喹啉和异喹啉的准确度和精密度(n=6)
3 结论
(1)建立了超快速液相色谱-串联质谱法同时测定婴幼儿纺织用品中喹啉和异喹啉残留的检测方法。(2)实验利用分散固相萃取对样品提取液进行净化,对分散吸附剂的选择,用量、吸附时间、解析时间以及解析液进行了系统优化。通过对方法基质效应的评价发现,所建方法基质干扰可以忽略,实验采用外标法定量。(3)喹啉和异喹啉的检出限均为0.16 μg/kg,加标回收率分别为90.2%~104.5%和91.6%~105.6%,相对标准偏差分别为2.4%~5.3%和3.8%~6.3%。该方法简单、快速、灵敏度高,稳定性好,能够满足婴幼儿纺织用品中喹啉和异喹啉的检测要求,同时也为其他纺织品中喹啉和异喹啉的监测提供技术支撑。
