根癌农杆菌Atu4856 基因原核表达载体的构建
2022-07-21刘啸宇后梦情程安琪胡传俊
刘啸宇,后梦情,程安琪,胡传俊,姚 坤,吴 萧
(宿州学院生物与食品工程学院,安徽宿州 234200)
农杆菌介导法是目前转化双子叶植物最为广泛使用的方法之一,该方法简单,转化机制明确、周期短,同时因转入基因的拷贝数少,因此大大地降低了因同源抑制而产生基因沉默的可能性[1]。同时,该技术具有转育周期较短、遗传性状较为稳定、变异幅度较小、对农艺和经济性状干扰较少等特点,已成为我国农作物进行外源基因导入时最常用的技术手段[2-3]。但同时也面临一些重要的技术问题,如转化效率低和转化结果的未知性和不可控性等。为了进一步了解根癌农杆菌T-DNA 转运的分子机制,探讨是否可以通过提高T- 复合物招募蛋白VBP 在农杆菌细胞内的表达量来提高T-DNA 的转化效率,这在植物转基因方面的应用具有重要意义。
1 材料与方法
1.1 试验材料
1.1.1 所用菌株
Agrobacterium tumefaciensC58,E.coliDH5α,实验室保存,菌液与70%的甘油等体积保存在冻存管中,冻存于-75 ℃冰箱中。
1.1.2 所用质粒
Prset-a,具有T7 启动子,实验室保存;pegfp-n1,含有egfp基因的质粒,实验室保存;prset-agfp1,Atu4856 基因上游495 bp 序列,带上标记基因gfp,插入启动子已除去的prset-a 质粒,构建。
1.1.3 所用引物
所使用的引物信息(划线部分为酶切位点) 见表1。

表1 所使用的引物信息(划线部分为酶切位点)
1.2 试验方法
1.2.1 用聚合酶链式反应扩增带有标记基因的Atu4856启动子序列
(1) 扩增495 bp 的启动子序列。以Pv1、Pv2 为引物,BamHⅠ酶切位点在引物Pv1 上,配制反应体系进行PCR 反应,结束后,进行琼脂糖凝胶电泳,并回收产物[4-5]。
(2) 聚合酶链式反应扩增标记基因——gfp。以Pgfp2,Pg2 为引物,以质粒pegfp-n1 为模板,BamHⅠ酶切位点在引物Pg2 上,待反应结束后,回收PCR产物,步骤同1.2.1 步骤(1)。
(3) 扩增Atu4856 启动子带标记基因的全长序列。将1.2.1 中(1) (2) 步骤的产物混合,以Pv1 和Pg2 为引物,配置反应体系进行扩增[6-7],产物回收,步骤同1.2.1 步骤(1)。
重叠延伸PCR 的过程和体系见图1,PCR 反应的体系见表2。

图1 重叠延伸PCR 的过程和体系

表2 PCR 反应的体系/μL
1.2.2 提取大肠杆菌prset- a 质粒
构建启动子探针载体的基本质粒prset-a 见图2。
1.2.3 PCR 扩增得到不含T7 启动子的prset- a 质粒
在T7 启动子基因序列的两端设计引物P1 和P2,均含有BamHⅠ限制性位点。同时,以prset-a质粒为扩增模板,从prset-a 质粒中去除T7 启动子,反应体系如表2 所述。PCR 反应结束后,PCR 产物回收步骤同1.2.1 步骤(1)。
1.2.4 DNA 的酶切、连接、转化及重组子的筛选
(1) 酶切以绿色荧光基因(gfp) 为报告基因的Atu4856 启动子序列。用BamHⅠ酶切含gfp的Atu4856 启动子序列。按照酶供应商提供的说明进行消化。按照表3 添加消化反应系统。消化反应体系置于37 ℃恒温水浴中水浴加热3 h。以非酶切序列为对照,用0.8%琼脂糖凝胶电泳鉴定酶产物,用试剂盒回收产物,并将其保存在-20 ℃条件下备用[8]。
酶切体系见表3。

表3 酶切体系/μL
(2) 去磷酸化和空载体酶切。用BamHⅠ酶切无T7 启动子prset-a 的PCR 产物。消化反应体系按表3混合,37 ℃下保温3.5 h。电泳后回收1% (m/V)琼脂糖凝胶,以防出现载体自连的现象[9],将按照表4 中体系进行载体去磷酸化。
载体去磷酸化体系见表4。
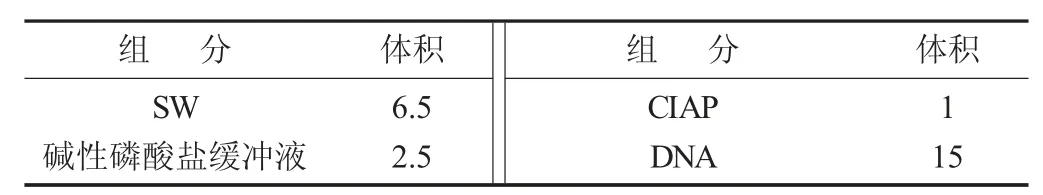
表4 载体去磷酸化体系/μL
(3) 连接。按照表5 所示的连接体系,将BamHⅠ酶切后的目的序列用T4 DNA Ligase 连接到BamHⅠ酶切后并去除磷酸化的载体上,于4 ℃下过夜连接,用于转化。
连接体系见表5。

表5 连接体系/μL
(4) 转化。将连接产物转化大肠杆菌DH5α 感受态细胞。
(5) 重组子的筛选。挑取单菌落,用LB 培养基37 ℃下培养,提取质粒,接着用BamHⅠ酶切、鉴定[10]。将与目的条带大小相同的阳性克隆片段进行测序,命名含有Atu4856 启动子的重组质粒,即prsetagfp1,并将此菌种进行保存。
(6) 检测探针载体的转录活性。挑取单菌落,用LB 培养基培养,在对数生长期时,对菌液进行离心、沉淀清洗,制片,在放大倍数为×400 的倒置荧光显微镜下观察是否有荧光。
2 结果与分析
2.1 带有gfp 基因的Atu4856 启动子序列扩增
以pegfp-n1 质粒为模板,经PCR 特异性扩增获得大小约750 bp 的绿色荧光蛋白基因(gfp) (图2)。设计搭桥PCR 引物,通过重叠延伸PCR,将gfp基因连接到Atu4856 启动子序列(图3),形成大小约1 225 bp 的嵌合基因片段,作为其是否具有启动子基因转录活性的报告基因。
PCR 扩增gfp基因电泳图见图2,PCR 扩增带有gfp的Atu4856 启动子电泳图见图3。
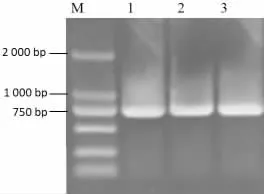
图2 PCR 扩增gfp 基因电泳图
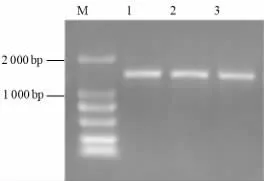
图3 PCR 扩增带有gfp 的Atu4856 启动子电泳图
2.2 PCR 扩增无T7 启动子的质粒(prset-a)
模板采用质粒prset-a,采用引物P1 和P2,通过PCR 扩增得到不含T7 启动子的质粒prset-a,大小约2.7 kb(图4),用于重组表达质粒的构建。
PCR 扩增去除T7 启动子的prset-a 质粒电泳图见图4。
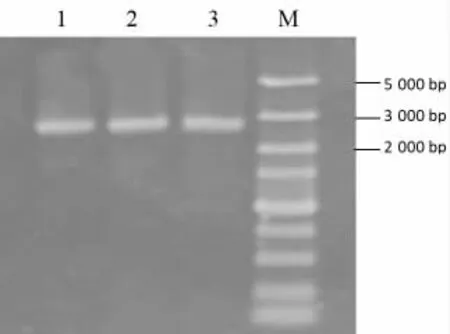
图4 PCR 扩增去除T7 启动子的prset-a 质粒电泳
2.3 酶切鉴定prset-agfp1 重组质粒
将BamHⅠ酶切后带有绿色荧光基因(gfp) 的Atu4856 启动子连接在BamHⅠ酶切后的prset-a 载体上,该载体无T7 启动子,接着转化到DH5α 大肠杆菌,培养过夜。经过提质粒、酶切、琼脂糖凝胶电泳等一系列过程,验证与PCR 产物一致(图5),同时将质粒测序,测序结果表明prset-a 质粒上成功插入了标记基因(gfp) 的Atu4856 启动子序列,载体构建成功。
酶切鉴定prset-agfp1 重组质粒电泳图见图5。
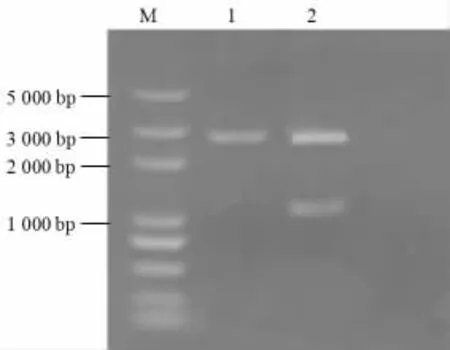
图5 酶切鉴定prset-agfp1 重组质粒电泳图
2.4 验证Atu4856 启动子在探针载体中的活性
取菌液制片,在倒置荧光显微镜下观察。结果显示,发射蓝光时有荧光,而只带prset-a 载体的大肠杆菌没有荧光现象(图6)。说明与gfp基因融合的载体已被成功构建,带有荧光基因(gfp) 的Atu4856启动子序列被证明具有转录活性。
未突变的含有495 bp Atu4856 上游序列重组质粒E.coli(prset-agfp1) 的荧光观察结果见图6。

图6 未突变的含有495 bp Atu4856 上游序列重组质粒E.coli(prset-agfp1) 的荧光观察结果
3 结论
启动子是构建基因表达载体的重要因素,对外源基因的表达水平具有着重要的影响。查阅大量文献、进行理论分析和试验工作,通过PCR 方法,获得了编码VBP3 蛋白Atu4856 基因的全长启动子序列。将prset-a 载体的T7 启动子与GFP 基因重组后,用Atu4856 启动子序列取代,成功构建了能在大肠杆菌中表达的启动子探针载体,并利用荧光蛋白研究农杆菌Atu4856 基因在大肠杆菌中的表达调控机制。
