复合催化剂改性矿用酚醛树脂低热值及发泡性能研究
2022-07-20胡相明祝富盛王其宝卢文欣余明高
胡相明,祝富盛,凌 青,王其宝,卢文欣,余明高,王 鹏,王 伟
(1.山东科技大学 安全与环境工程学院,山东 青岛 266590;2.山东科技大学 矿山灾害预防控制省部共建国家重点实验室培育基地,山东 青岛 266590;3.山东科技大学 化学与生物工程学院,山东 青岛 266590;4.重庆大学 煤矿灾害动力学与控制国家重点实验室,重庆 400044;5.河南理工大学 安全科学与工程学院,河南 焦作 454003;6.中煤科工集团沈阳研究院有限公司 煤矿安全技术国家重点实验室,辽宁 抚顺 113122;7.煤炭科学研究总院,北京 100013)
0 引 言
煤炭是世界范围内重要的能源,支撑着全球经济和社会发展,但煤炭在生产、存储和运输过程中发生的自燃现象是世界性难题[1]。煤炭自燃往往与煤层厚度、煤质、地质结构有关,是煤层氧化产热大于环境散热的结果,限制环境温度和堵漏风减少氧气向煤层扩散被认为是防治煤自燃的重要技术手段[2]。目前矿用堵漏风材料主要有无机材料和有机材料2大类[3],其中无机材料主要有发泡水泥、复合胶体、石灰-石膏-纤维复合材料等材料,有机材料主要是有机聚合物的发泡材料,如聚氨酯泡沫、酚醛树脂泡沫等。无机材料存在密度较大、发泡困难的缺陷,且充填后不能堆积结顶,限制了其使用;有机材料如聚氨酯、酚醛树脂等的易发泡特性使其在保温材料工业中被广泛使用,但也存在易燃的问题。相比之下,酚醛树脂难燃且具有抗高温岐变、高温释放烟气低等优点,这使其成为目前矿井内最常使用的堵漏风泡沫的原料[4]。但是,目前的工艺中,酚醛树脂泡沫存在发泡固化后温度过高的问题,发泡固化后的树脂泡沫体超过100 ℃,而过高的温度会导致酚醛树脂泡沫体的热量积聚传导至煤层,增加煤炭自燃的风险[5-7]。降低酚醛树脂在发泡过程中的固化放热量,降低酚醛树脂发泡固化后的泡沫体温度成为防治矿区煤自燃的首要任务。
酚醛树脂固化发泡过程的放热主要来源分为2个部分:① 羟甲基和酚羟基的交联反应放热,这与树脂中羟甲基的数量相关,羟甲基数量越多,缩聚反应越快,热量越高;② 碱性催化剂与酸性固化剂中和反应放出的热量,碱性越强的催化剂,中和时放热越多。降低树脂固化温度,就要降低树脂的羟甲基化程度或改变酚醛树脂合成中的碱或发泡过程中的酸的成分,并及时将反应释放的热量排出泡沫体外,延缓热量积累[8-10]。目前的研究大多关注降低发泡过程的反应热,研究工作主要集中在改变羟甲基化程度和酸性固化剂的复配领域,如王兆华等[11]用氯化苄改性酚醛树脂,在酚醛树脂中引入苄醚基,使得苯环上的邻对位活性和羟甲基活性降低,通过降低树脂中羟甲基总量来降低树脂的固化温度,提高了酚醛树脂的贮存期;刘欣等[12]通过对各类酸性固化剂的复配,确定固化剂中的酸强度对固化放热影响明显,发泡体的最高温度随着磷酸和硫酸用量的增加呈现明显的升高趋势。对于参与中和反应的碱性催化剂并没有引起研究人员的重视,树脂的合成过程也往往认为与发泡过程的放热无关。但碱性催化剂是影响苯酚活性的关键,能够影响树脂合成过程中的羟甲基化程度,同时影响与酸性固化剂的中和反应程度,间接影响固化过程的放热时间。如能拉长树脂在固化过程中的放热时间,就能给予发泡体足够的热量散失时间[13-15],使得发泡固化后泡沫体的温度降低。
因此,笔者团队从催化剂入手,采用自制的催化剂A,制备了羟甲基化程度低于普通酚醛树脂的改性酚醛树脂N-PF,延长了储存时间的同时减少了发泡过程的放热量,发泡后的泡沫体较一般酚醛树脂发泡体温度降低46%。通过对比不同碱性催化剂对树脂黏度、固含量、泡沫固化温度和发泡倍率的影响,采用扫描电镜技术从微观层面分析了合成的酚醛树脂改性后的化学结构的变化以及发泡后的物理形貌。结合DSC曲线和动力学方程证明,改性酚醛树脂与酚醛树脂的固化过程均为一级反应。试验证明,催化剂A通过降低苯酚邻对位的电子云密度,减少了羟甲基指数,使改性酚醛树脂在发泡固化过程中的总放热量减少,放热时间延长,两者共同作用大幅度降低了改性酚醛树脂发泡固化后泡沫体的温度,减少了对环境的热量释放。
1 改性酚醛树脂材料与方法
1.1 试验材料
苯酚、多聚甲醛、碱性催化剂(氢氧化钾、氢氧化钠、碳酸氢钠、氨水、碳酸钙)、固化剂(硫酸、磷酸、对甲苯磺酸(PTSA))、表面活性剂(吐温-80)、发泡剂(正戊烷)等药品均为分析纯,购自成都市科龙化工试剂厂,催化剂A为实验室自制复配,矿用发泡材料物料B购自河北省邢台茂森聚氨酯有限公司。
1.2 树脂材料的制备及结构分析
1.2.1 树脂制备
在装有温度计、球形冷凝管、机械搅拌器、恒压分液漏斗的四口烧瓶中加入一定量的苯酚,将四口烧瓶放入恒温水浴锅升温至80 ℃,恒温5 min后加入1.8倍物质的量的多聚甲醛(分3批次投入,每次间隔40 min),同时投放催化剂充分搅拌,反应120 min后冷却至室温出料。不同催化剂获得的酚醛树脂分别进行表征(图1)。
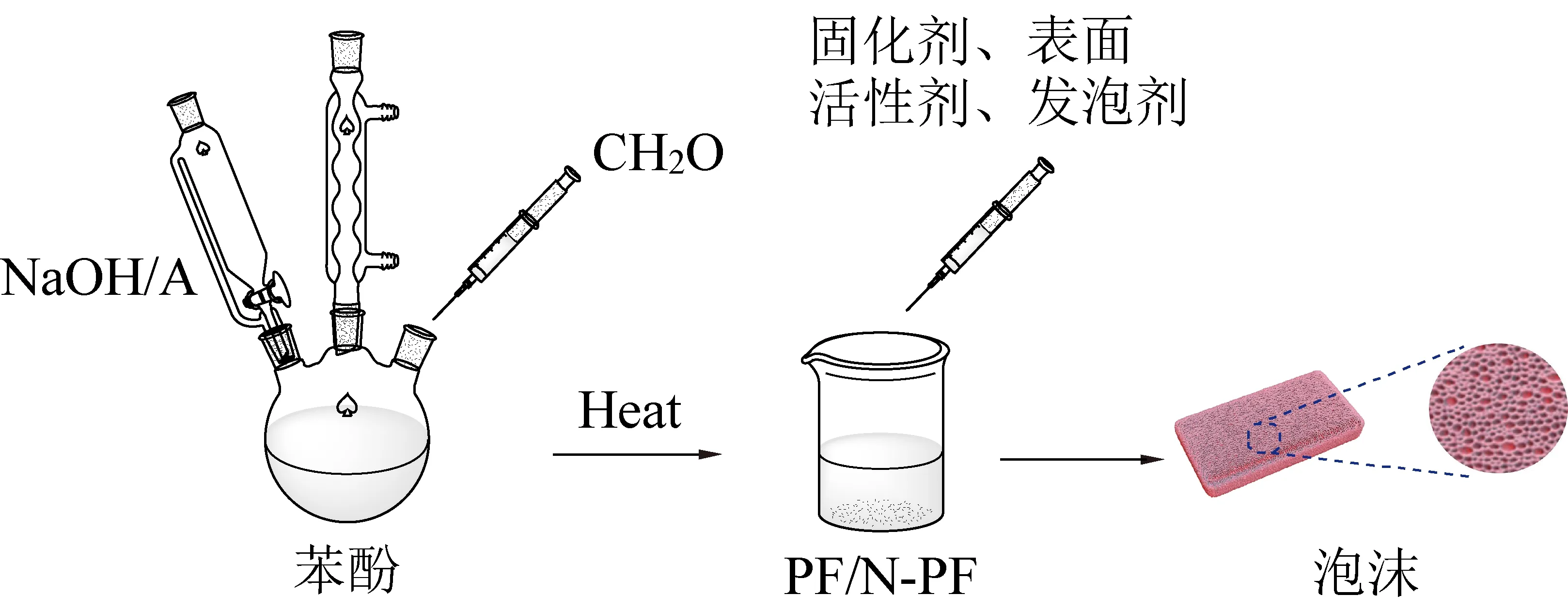
图1 酚醛树脂的合成Fig.1 Synthesis of phenolic resin
1.2.2 树脂发泡材料制备及发泡温度测定
室温下,在多个相同规格的250 mL锥形瓶中加入相同质量(32 g)的PF或N-PF。为保证试验的准确性,固化剂、发泡剂和表面活性剂统一使用相同质量的发泡材料物料B。发泡试验开始时将相同质量(8 g)的矿用发泡材料物料B倒入锥形瓶,以酚醛树脂质量为基准,其他组分按酚醛树脂的质量百分比加入,加入物料B后快速搅拌,塞上插入TM902C型热电偶的橡胶塞,使探头距锥形瓶底部3 cm,监测密闭容器最高温度,记录热电偶最高温度。同时使用秒表记录固化时间,树脂内部温度显著下降时终止记录。
1.3 结构分析
红外光谱(FT-IR)分析:采用Nicolet IS50型傅里叶变换红外光谱仪,其中树脂采用溴化钾压片方式测量,酚醛泡沫切片后采用表面全反射(ATR)附件进行测试。
核磁碳谱分析:使用氘代二甲基亚砜(DMSO-d6)溶解样品,采用JEOL JNM-ECZ 400S型核磁共振波谱仪进行液态核磁信号的收集和分析,对核磁碳谱信号采用MestNova软件进行处理,以ppm为化学位移单位获得核磁分析图。
DSC分析:采用瑞士Mettler DSC1型差示扫描量热仪对所合成的酚醛树脂进行表征,升温范围25~300 ℃,N2保护,流量40 mL/min。
SEM分析:使用APREO型扫描电子显微镜,从泡沫表面获取SEM图像。为增强样品的导电性,在测试之前将样品进行镀金处理。
2 结果与讨论
2.1 不同碱性催化剂对树脂黏度和固含量的影响
工业上黏度过大的树脂无法发泡,树脂是否失效往往可以用树脂黏度进行判断,黏度越高聚合度越高,其固含量也相应增加,与发泡剂反应而发泡的难度越大。首先以不同种类的碱性催化剂(氢氧化钠、氢氧化钾、氨水、碳酸氢钠、新型催化剂A和碳酸钙)为考察对象设计单因素试验,以酚醛树脂的固含量和黏度作为树脂合成的考核指标。
如图2所示,树脂的黏度存在氢氧化钠>A>氢氧化钾>碳酸氢钠>氨水>碳酸钙的规律,与催化剂的碱性成正比,碱性越强树脂黏度越高,原因在于碱性越强,对酚醛缩聚反应的催化效率越高,在相同时间内交联所得的产物分子量相对更大;而氨水的碱性略高于碳酸氢钠,但碳酸氢钠合成的酚醛树脂黏度高于氨水催化合成的酚醛树脂,其原因在于氨水中直接以氢键缔合的水分子,在整个酚醛树脂交联过程中,与树脂充分混合,降低了树脂的黏度。
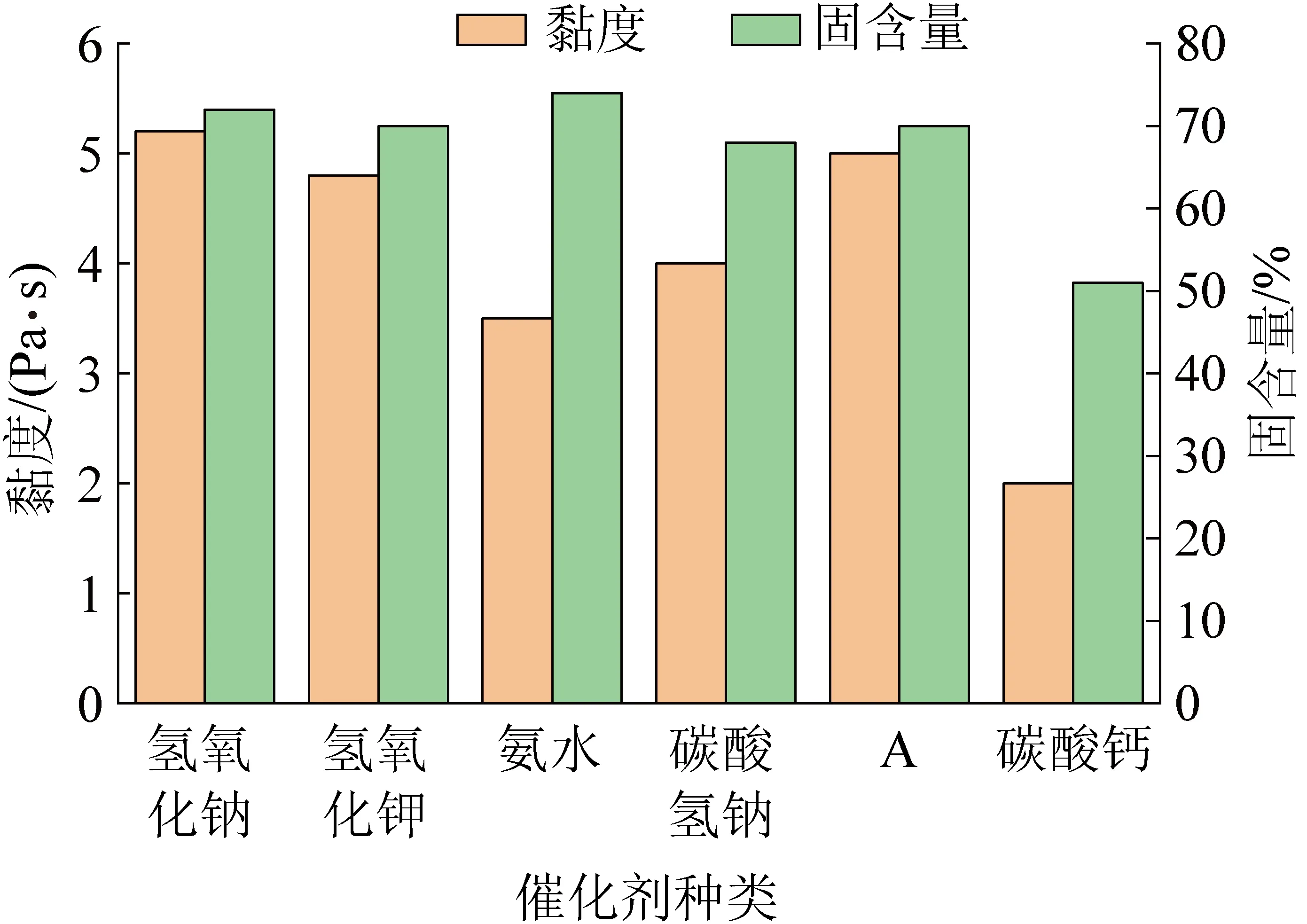
图2 不同碱性催化剂对树脂黏度和固含量的影响Fig.2 The effect of different alkaline catalysts on viscosity and solid content
同样,树脂中的固含量存在氨水>氢氧化钠大于氢氧化钾>A>碳酸氢钠>碳酸钙的关系,碱性高固含量相对更高,但氨水显示出特殊的位次,这是由于氨水受热后易于挥发,导致酚醛树脂中的游离酚、游离醛和水在高温烘干条件下蒸发而浓缩;同时,氨水在酚醛缩聚反应中产生含氮中间体与甲醛反应形成烯胺中间体,导致酚醛树脂的交联程度较大,产物呈现出异乎寻常的高固含量。
综上,与最常用的氢氧化钠催化合成的酚醛树脂相比,催化剂A催化合成的酚醛树脂的黏度、固含量等均略小,其原因是其复合结构导致碱性释放缓慢,对苯酚的活化作用缓慢,催化的反应活性位点更少,导致缩聚反应所得产物分子量适中,其黏度和固含量都表现出较为适宜的发泡条件。
反应机理的研究认为,碱性下(如氢氧化钠)酚醛树脂的合成是首先形成苯酚负离子,受p-π共轭作用影响,苯酚的氧负离子向苯环迁移电子,并在苯环氧原子的邻对位形成了富电子区。随后,甲醛碳原子的碳正离子与苯环富电子区结合,形成羟甲基苯酚,并逐步进行下一步反应。催化剂A的碱性弱于氢氧化钠,强于碳酸氢钠,这导致催化剂A催化下的苯酚中苯环电子云密度不如氢氧化钠催化的苯酚,最终形成的酚醛树脂羟甲基指数下降,这一点与后续的核磁及红外表征一致,合成机理如图3所示。
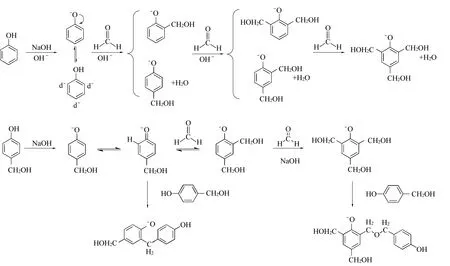
图3 酚醛树脂合成机理Fig.3 Mechanism of phenolic resin prepolymer
2.2 不同碱性催化剂对泡沫固化温度和发泡倍率的影响
为测定不同催化剂对所合成的酚醛树脂在发泡过程中的固化温度和发泡倍率带来的影响,我们进一步对所合成的酚醛树脂进行发泡试验,以32 g酚醛树脂获得的酚醛树脂发泡体的固化温度和发泡倍率作为对催化剂在发泡过程中的筛选考核指标。室温下称取,发泡条件保持相同(表1)。
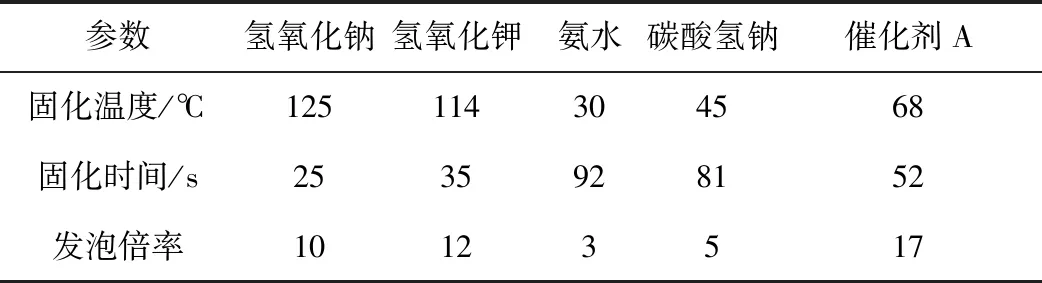
表1 不同催化剂合成树脂考察指标 Table 1 Investigation index table of synthetic resins with different catalysts
相对于催化剂对黏度和固含量影响的程度,不同催化剂合成的酚醛树脂在发泡阶段出现了出乎意料的差异性,如图4所示,酚醛树脂发泡体固化温度与催化剂碱性强弱呈现良好的线性。氢氧化钠碱性最强,其催化合成的树脂固化温度最高,固化时间最短,短时间内集中放热程度最高;氢氧化钾次之,催化剂A、碳酸氢钠依次降低。氨水再次呈现出异常,原因在于其较低的黏度导致发泡容易,同时氨气的释放利于温度降低。
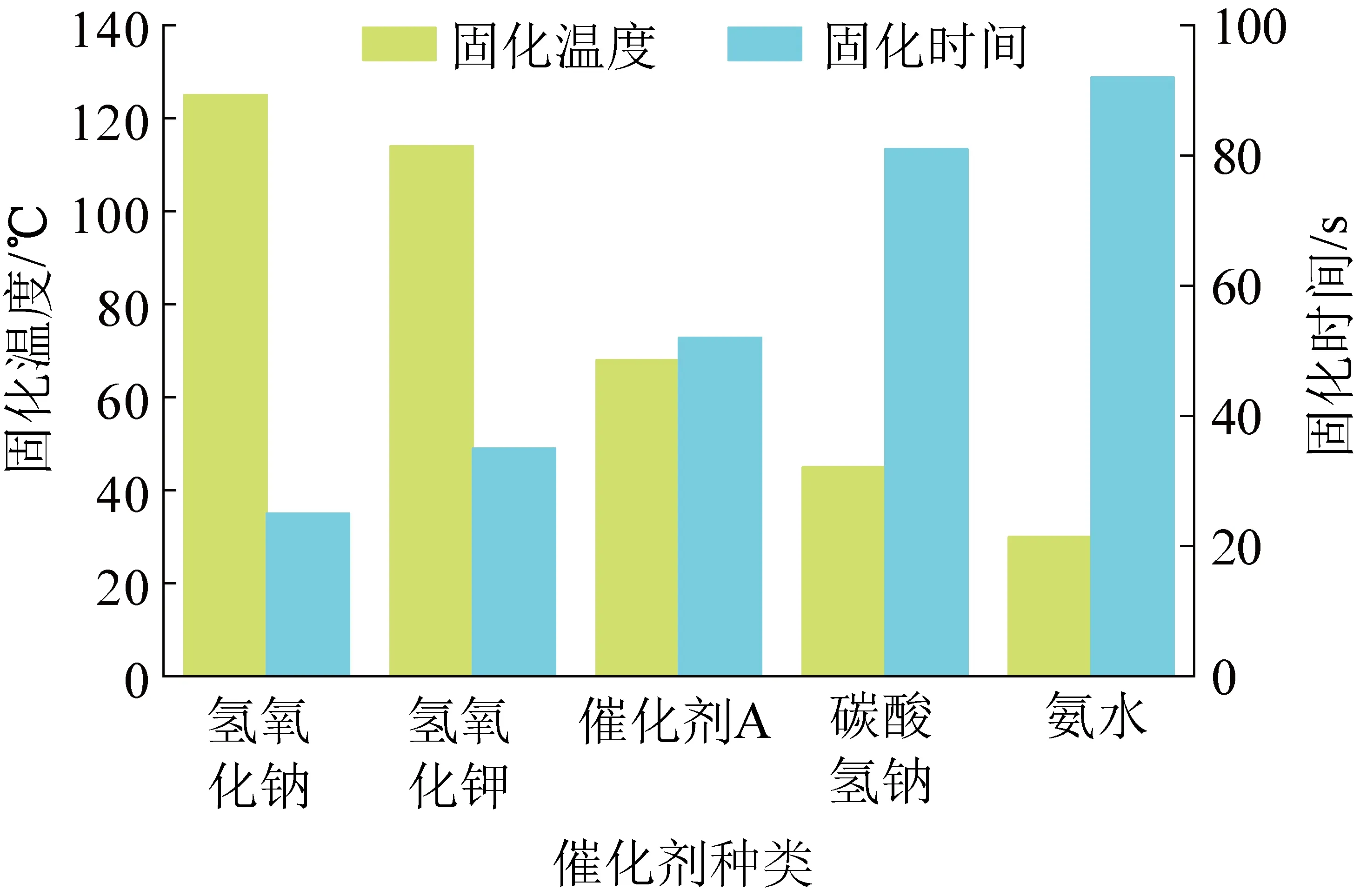
图4 不同催化剂合成树脂的固化温度和固含量对比Fig.4 Curing temperature and solid content of synthetic resin with different catalysts
除氨水外,其他强碱性催化剂合成的酚醛树脂固化温度高的原因是,发泡过程中添加的酸性固化剂与强碱性催化剂酸碱中和释放出大量的热,进而加快了酚醛树脂初始阶段的交联反应,使树脂中活性位点反应加速,过快的固化速度使得热量集中释放造成热量聚集和固化温度的升高。
同样由表1可知,催化剂碱性的强弱对发泡倍率的影响明显,氢氧化钠碱性最强,固化速度最快,树脂固化速度过快导致树脂发泡膨胀的过程缩短,使得氢氧化钠合成的树脂发泡倍率低于氢氧化钾;当催化剂碱性过低时,树脂的交联速度变慢,在发泡过程中不能持续提供热量,固化反应速率降低,树脂难以及时固化,酚醛树脂发泡后的泡孔结构难以维持造成塌泡,树脂的最终发泡倍率降低[16]。
从对比试验能够看出,与通常采用的氢氧化钠催化剂相比,催化剂A催化合成的酚醛树脂能够大幅度降低发泡体中心温度,同时不影响其发泡倍率等性能指标。与其他研究者针对替换单体改性酚醛树脂的方法相比,改变催化剂合成酚醛树脂能够在不增加成本的基础上,获得储存时间长、发泡固化后泡沫体温度更低的改性酚醛树脂堵漏风材料,这对于煤矿安全生产和经济效益具有极高的价值。
2.3 正交试验设计
为在机械性能不降低的情况下尽可能降低酚醛树脂的固化温度,进一步对催化剂A合成的酚醛树脂的发泡工艺进行了优化,以期获得最适宜的发泡配方和发泡条件。依据现有研究中复配固化剂、发泡剂、表面活性剂3个因素对树脂固化温度、发泡倍率具有较大影响的特点,结合催化剂A的影响,我们选取7个因素A~G:催化剂A、吐温-80、正戊烷、PTSA、硫酸、磷酸及水的用量进行正交试验并设计正交试验因素水平表(表2),以树脂固化温度和发泡倍率作为性能指标进行考察[17]。
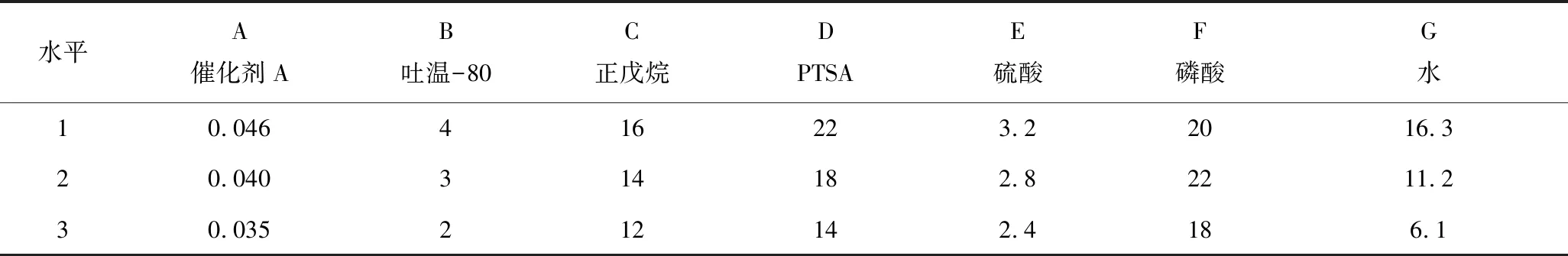
表2 试验因素水平Table 2 Factor-level values of orthogonal test %
本试验为7因素3水平的正交试验,在不考虑交互作用的基础上,按照正交试验规定,需选用L18(37)型表格来安排试验。
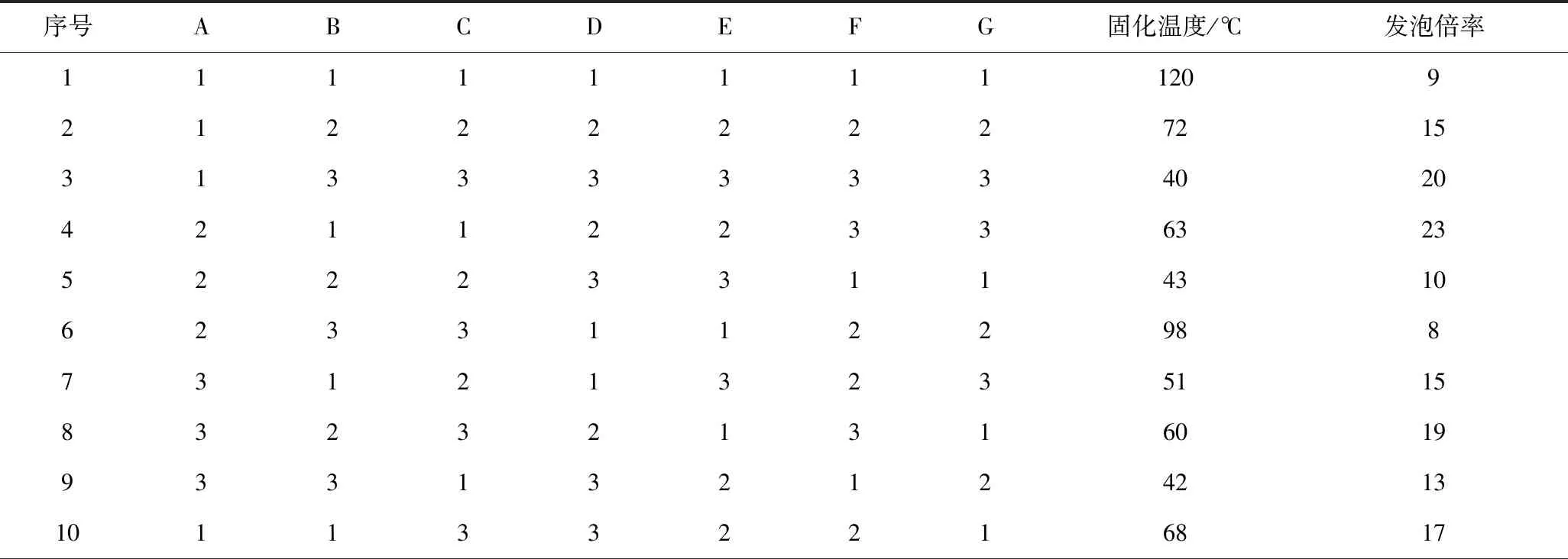
表3 正交试验结果Table 3 The orthogonal experimental results
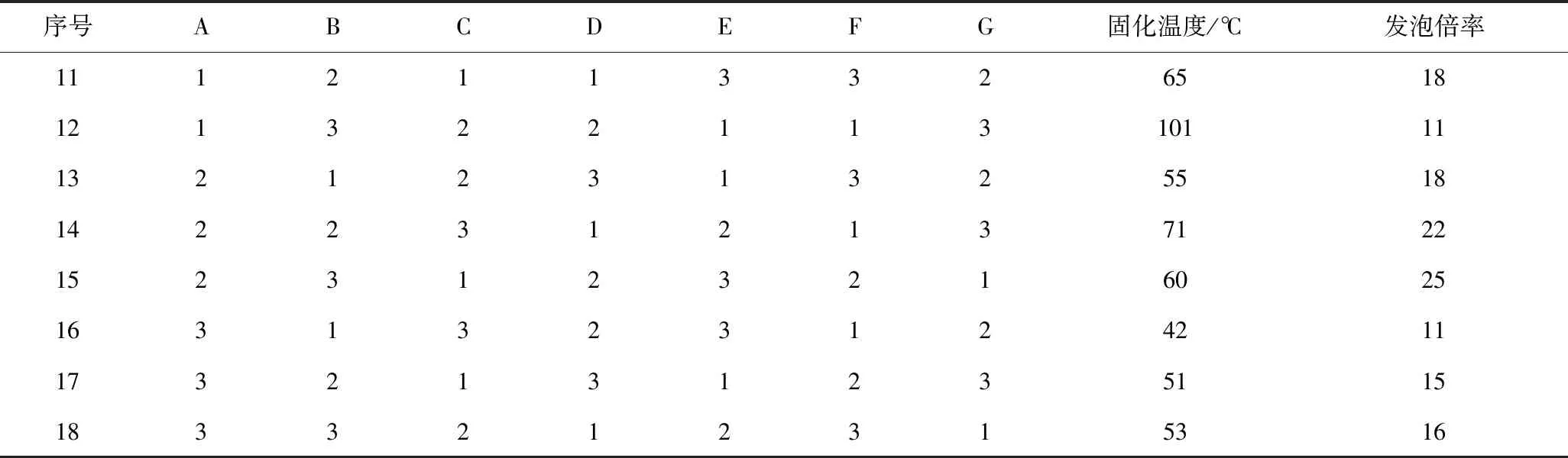
续表
由极差分析可知,影响固化温度的因素排序为:硫酸>催化剂A>PTSA>磷酸>吐温-80>水>正戊烷,以固化温度和发泡倍率为指标所得最优方案结果并不一致,因此对所选因素和水平进行适当调整,由表4可以看出,PTSA、硫酸、磷酸和催化剂A的固化温度都会随着用量的增加随之升高,进一步验证了树脂发泡固化的过程中,酸碱中和反应放热加速了集中放热的历程。相同的是,PTSA、硫酸和催化剂A酚醛泡沫在固化过程中,随着用量的增加,发泡倍率呈现先增大后减小的趋势,这是因为过弱的酸碱中和放出的热量难以将致密的泡孔结构固定在泡沫体内,造成塌泡现象的产生,而过高的酸碱性导致中和反应释放出过高的热量,在泡沫体尚未形成均匀泡孔结构前发泡过程就已结束,造成泡孔结构过于致密。考虑矿井充填堵漏应用中需要合适的发泡倍率,选用A2,D2,E2作为最优方案较为合适。磷酸由于其中强酸的特性,发泡倍率随着用量的增加随之增加,因此选择F3作为最优方案;水、吐温-80和正戊烷对固化温度及发泡倍率影响较小,根据极差分析表格和环保角度考虑,选用G1,B2和C3作为最优方案。
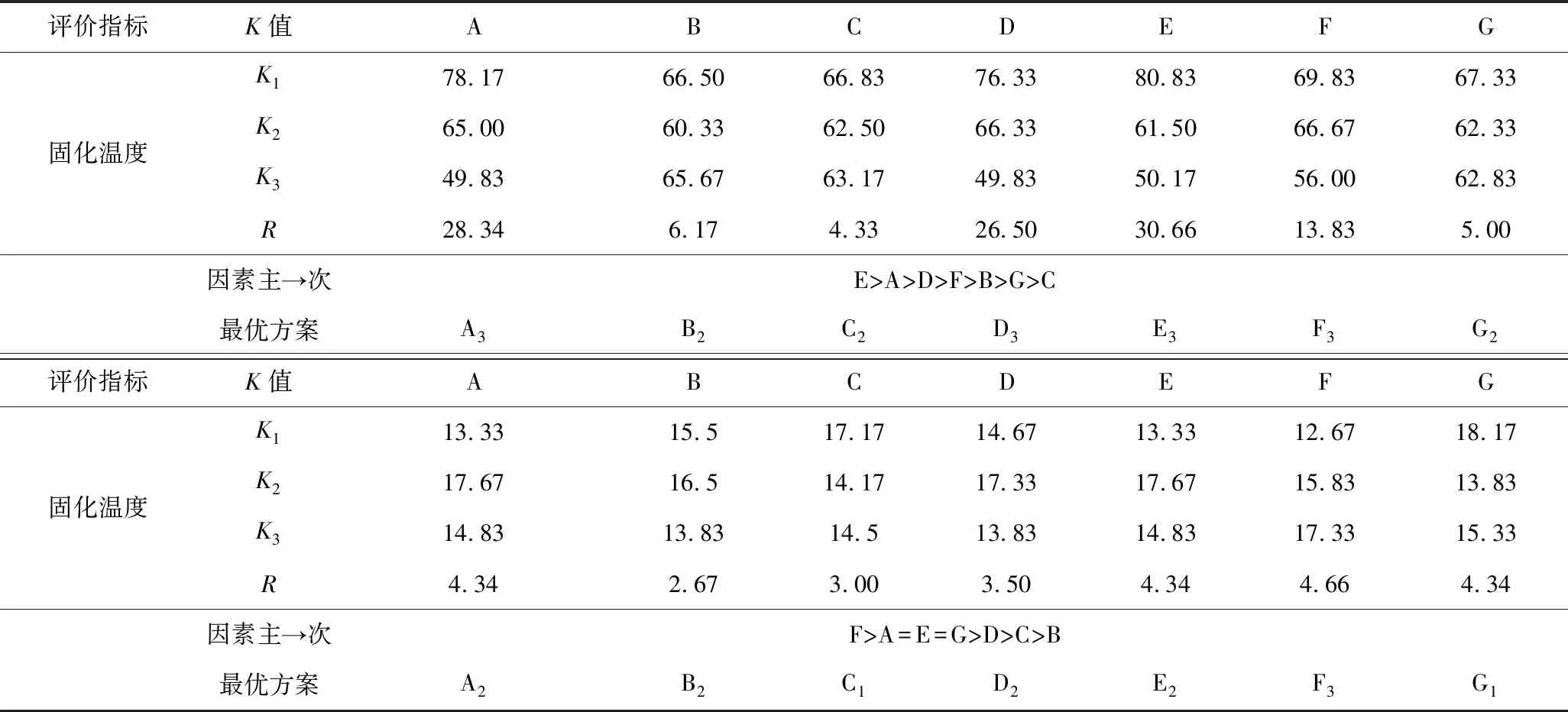
表4 试验结果分析Table 4 Analysis of test results
2.4 FT-IR分析

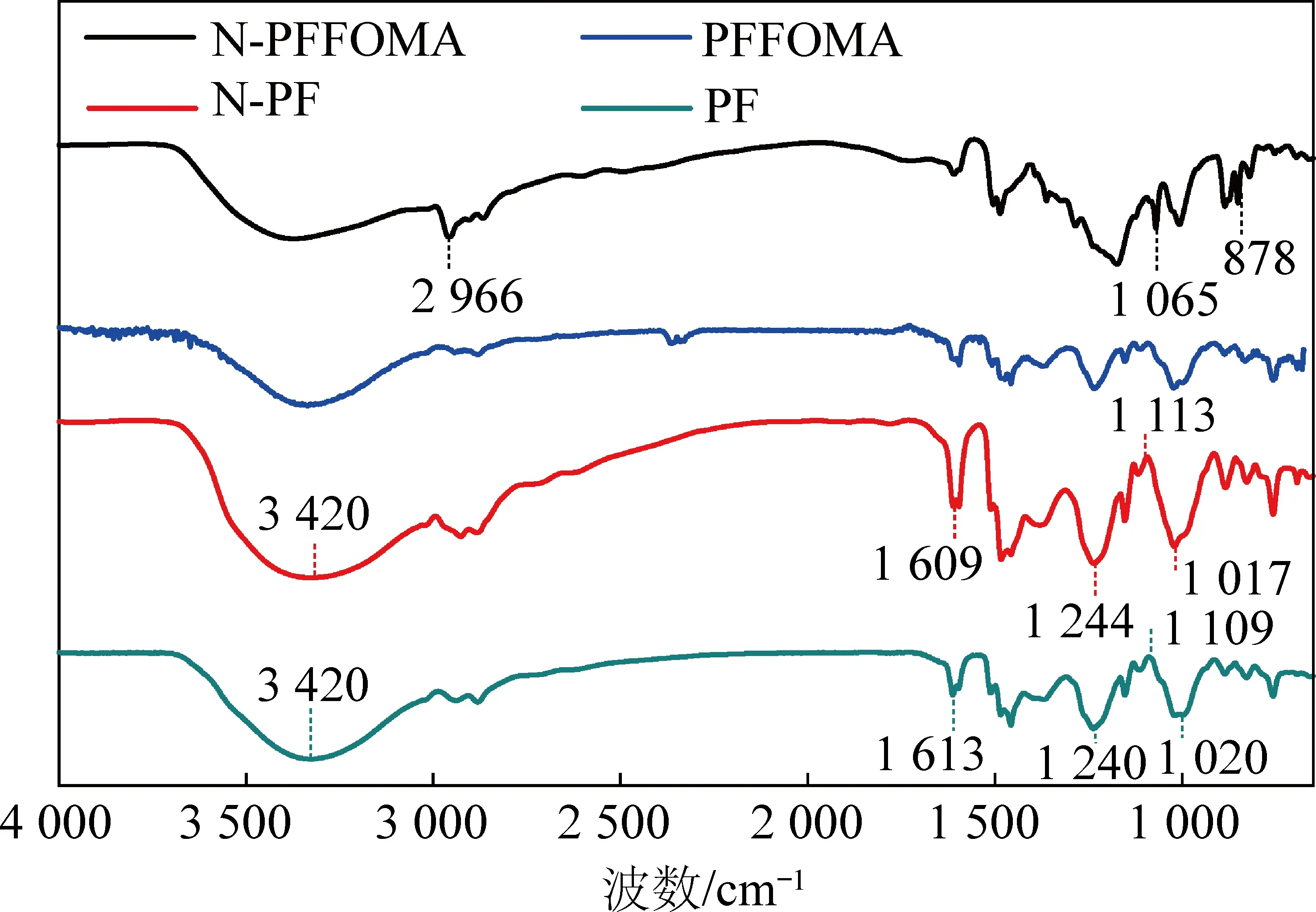
图5 PF和N-PF的红外谱图Fig.5 FT-IR spectra of PF and N-PF resins
2.5 核磁碳谱分析
图6和7为氢氧化钠合成的PF和催化剂A合成的改性N-PF的13C-NMR谱图,可以得知,PF与N-PF化学结构主体相同,a区域δ=151~157,是苯环上连接酚羟基的碳质子峰;b位置δ=115~132处是除链接酚羟基外,苯环上其他碳的质子峰;c处δ=71.61和δ=68.98处为醚键—CH2—O—CH2—的碳质子峰;d处δ=64.25~64.60处为链接苯环上的—CH2OH的羟甲基碳质子峰;δ=39~40处为溶剂氘代二甲基亚砜的化学位移。由图6可以看出,催化剂A催化的树脂在b区域向δ=115处偏移,这是由于催化剂A的碱性降低,苯酚去离子化趋势减缓,苯环上碳原子受酚羟基负离子的供电子效应相应降低,导致反应中的活性下降,化学位移向低场区偏移。活性下降的结果导致树脂中苯环间形成的连接结构(苯甲基醚、苯甲基酮等)数量与刚性均相对降低,最终获得的N-PF泡沫的脆性相应降低。
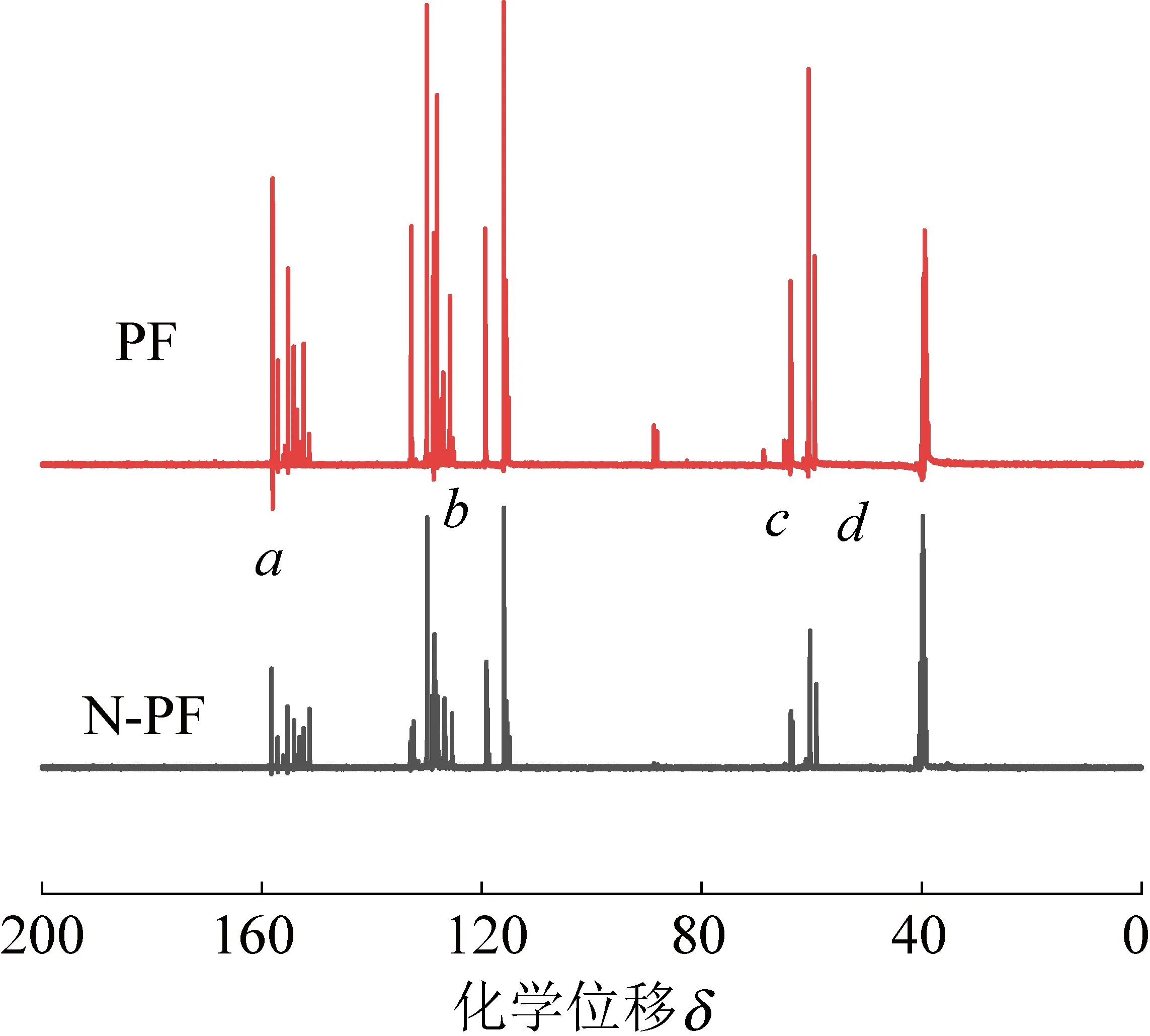
图6 PF和N-PF的13C-NMR谱图Fig.6 13C-NMR spectra of PF and N-PF
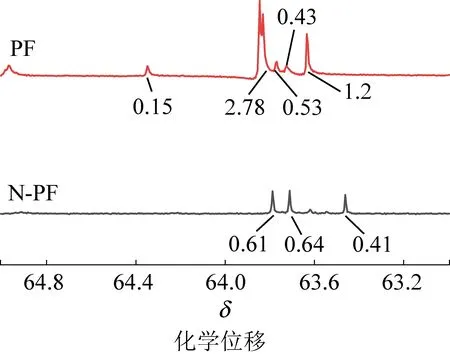
图7 PF和N-PF的13C-NMR谱图(δ=63~65)Fig.7 13C-NMR spectra of PF and N-PF(δ=63-65)
固化温度通过影响树脂的羟甲基化程度,从而影响树脂在后续固化过程中缩聚反应的速率。PF和N—PF的—CH2OH中碳吸收峰在13C—NMR上的δ=62~65区域,13C—NMR如图4所、图5所示,分别采用在128.58处的苯环上不参与反应的碳质子峰作为内标,设定其峰面积为1.00,分别对PF和N—PF在—CH2OH处的碳质子峰的峰面积进行积分,结果显示,PF的峰面积为5.09,N-PF为1.66,说明PF的羟甲基总量明显高于N-PF。羟甲基是交联过程中最重要最活泼的基团,改性N-PF中的羟甲基总量更低,充分说明了催化剂A所合成的酚醛树脂的发泡活性点的降低,与固化剂反应时具有更低的反应活泼性,导致反应的放热趋于平缓。
根据反应热力学,反应放出的总热量与反应的化学键及总量有关,因此不同催化剂合成的酚醛树脂的发泡过程的放热总量相似,温度变化大的原因主要与放热时间有关,即影响温度变化的关键因素其实是固化过程的时间,时间越长,发泡体对外释放的热量越多,发泡体内部温度就越低。为验证这一推测,进一步使用差热扫描分析对树脂固化过程的放热情况进行了分析。
2.6 SEM分析
为对比分析改性酚醛树脂N-PF与PF泡沫的微观结构,使用扫描电子显微镜放大150倍时对材料泡孔结构进行观察,如图6所示。通过孔径测量发现,N-PF泡沫的孔径在300~400 μm,显著高于PF泡沫的孔径(110~140 μm),说明新型改性酚醛树脂N-PF显著提高了树脂的发泡倍率,同时由图8可以明显看出,PF泡沫有大量碎屑。本文采用同样的方法制备PF及N-PF泡沫样本,但是在PF样本表面有大量的碎屑,相反N-PF泡沫表面没有碎屑,由此可以说明N-PF泡沫粉化率低于PF泡沫,说明N-PF泡沫的力学稳定性方面表现出更优异的性能。

图8 N-PF泡沫和PF泡沫的SEM图Fig.8 SEM diagrams of N-PF foams and PF foams respectively
2.7 固化动力学分析
2.7.1 DSC与固化温度
为研究催化剂A对树脂固化过程的影响,分别以5,10,15和20 ℃/min的升温速率对N-PF和PF树脂的DSC曲线进行分析,如图9、图10所示。不同升温速率下,PF和N-PF表现出相同的放热规律:随着升温速率的增大,放热峰向高温方向偏移,放热面积相应增大,即放热焓变增大;随着升温速率的增大,峰始温度(Ti),峰顶温度(Tp)和峰终温度(Te)均向高温方向偏移导致固化时间缩短。造成这些现象的原因在于低升温速率下,固化体系有充足的时间进行固化反应,而高升温速率的条件下,体系中的分子链来不及进行规则排列,造成放热集中,固化温度升高[19-21]。
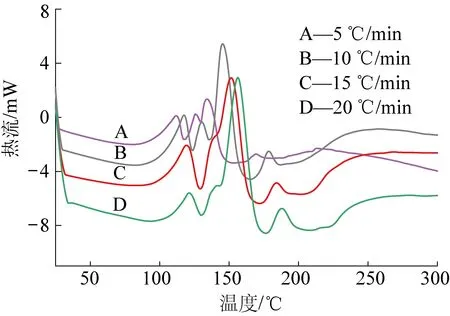
图9 不同升温速率下N-PF的DSC曲线Fig.9 DSC curves of N-PF resin at different heating rate
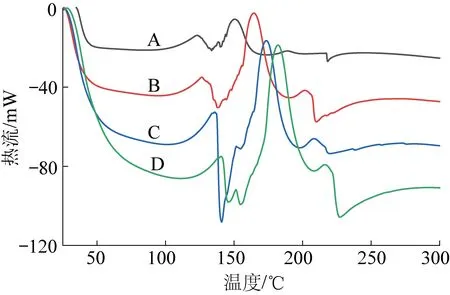
A—5 ℃/min,B—10 ℃/min,C—15 ℃/min,D—20 ℃/min图10 不同升温速率下PF的DSC曲线Fig.10 DSC curves of PF resin at different heating rate
相同地,如图11所示,对比相同升温速率下PF和N-PF的固化行为,PF的放热峰,Ti,Tp,Te均表现出较N-PF高温方向移动的趋势,这就说明了在相同升温速率下,PF的热惯性大于N-PF,导致PF与N-PF前后温度差距扩大,因此固化放热峰出现在相对更靠后的位置。见表5,在相同时间内,N-PF放出的热焓变仅为PF放热焓的1/2,固化时间却为PF的2倍,由此可以得出N-PF的固化过程中,催化剂A能够起到缓释作用,热量有足够的时间逃逸出泡沫体外,造成固化温度低的现象,这一点与核磁碳谱的分析结果一致。
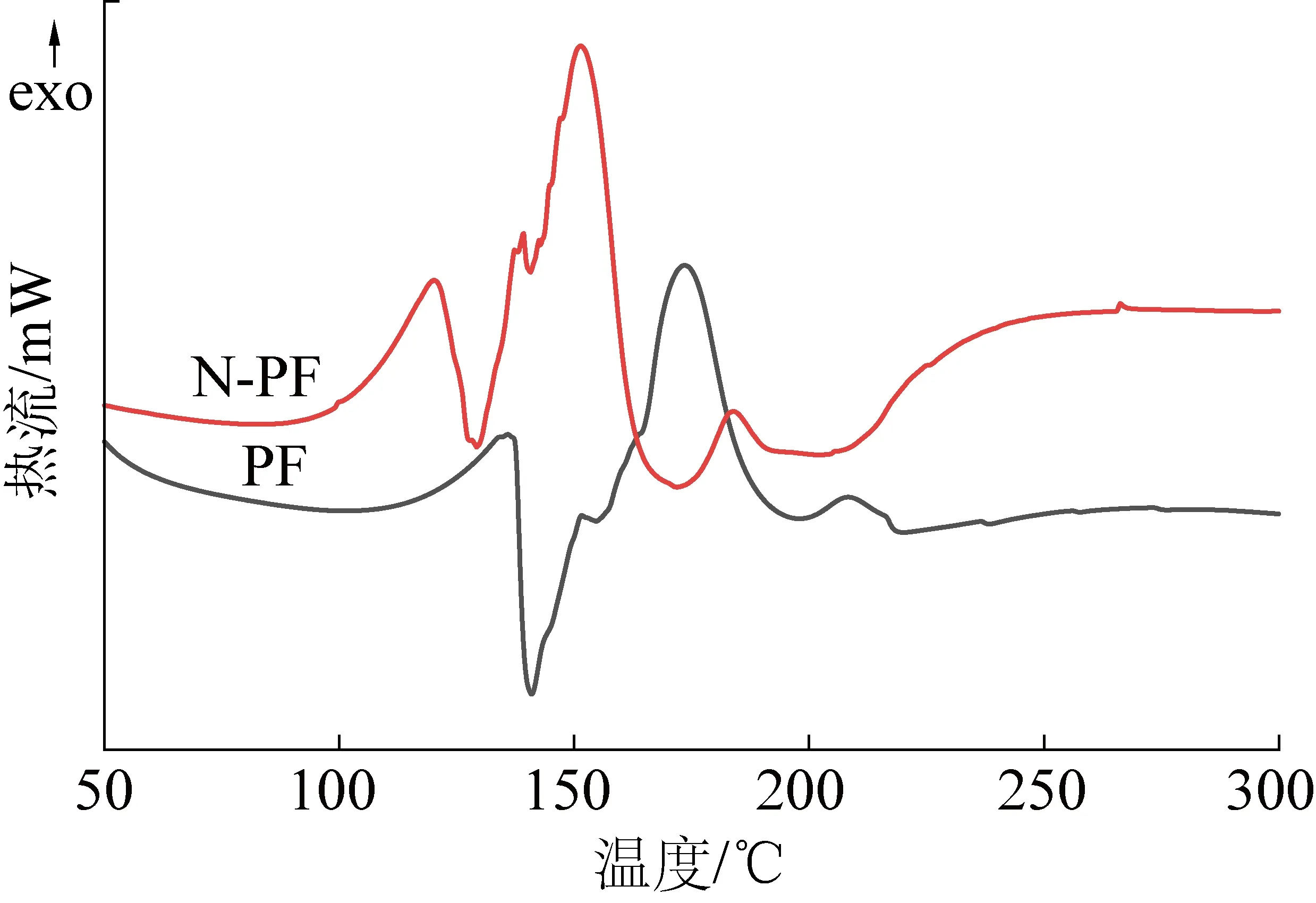
图11 PF和N-PF的DSC曲线(升温速率15 ℃/min)Fig.11 DSC curves of PF and N-PF resin Heating (rate 15 ℃/min)
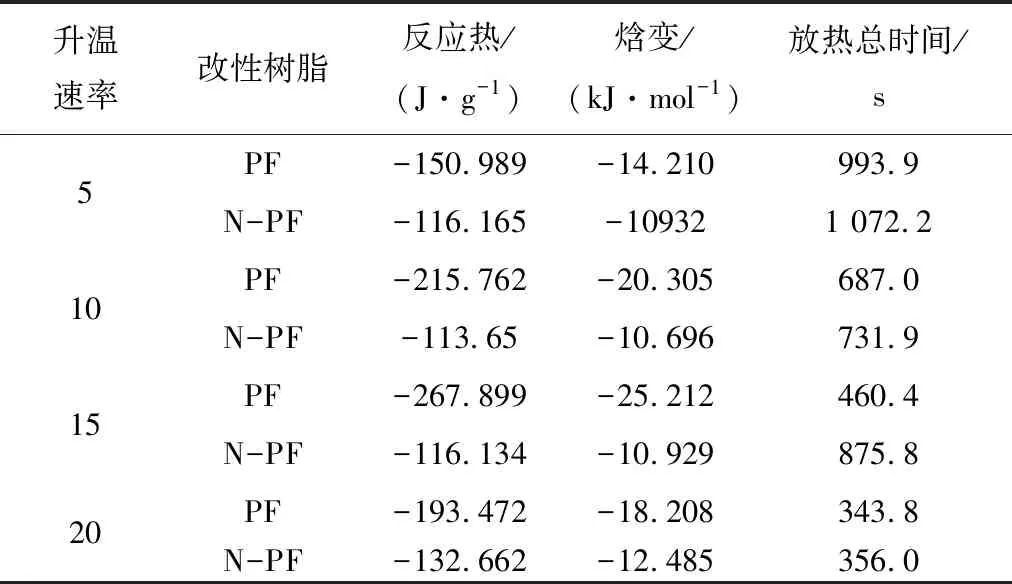
表5 PF和N-PF的DSC参数Table 5 DSC parameters of PF and N-PF
为进一步定量表征固化反应过程,采用热分析法对PF和N-PF的固化过程建立反应动力学模型,计算出固化动力学参数,明确催化剂A对固化过程的具体影响。
2.7.2 固化动力学方程的建立
固化反应的活化能随着反应进程逐渐增大,采用Kissingger法进行动力学分析[22-24],所得结果即为整个体系的表观活化能,Kissinger方程如式(1)所示:
(1)
其中,Φ为升温速率,℃/min;Tp为放热峰值温度,K;A为指前因子;R为理想气体常数,R=8.314 J/(mol·K);Ea为反应活化能。
按照Kissinger方程进行不同升温速率下DSC测试,由表6中的数据绘制曲线,如图12所示,利用回归计算可求得活化能Ea和指前因子A。
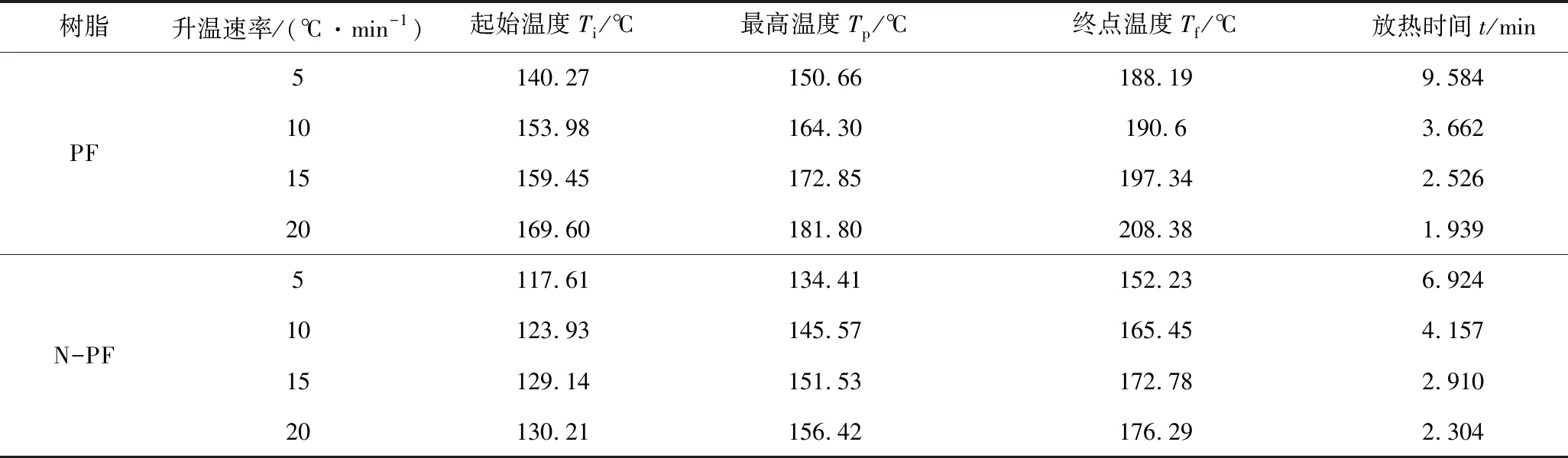
表6 不同升温速率下PF和N-PF体系固化反应的特征参数Table 6 Characteristic parameters of the curing reaction of PF and N-PF system at different heating rate
相同的,采用Crane方法来确定反应级数,Crane方程如式(2)所示:
(2)
由表7数据绘制曲线,如图12所示,根据斜率代入式(3)即可求得n。由图12、图13可知,两条曲线有很好的线性关系,所得样品固化动力学参数见表7。
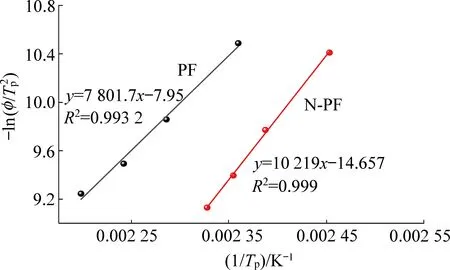
图12 使用Kissinger方程拟合实验数据Fig.12 Fitting of the experimental data using the Kissinger equation
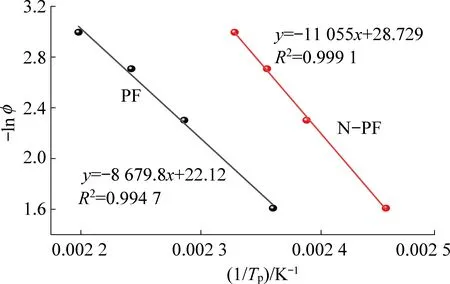
图13 使用Crane方程拟合试验数据Fig.13 Fitting of the experimental data using the Crane equation
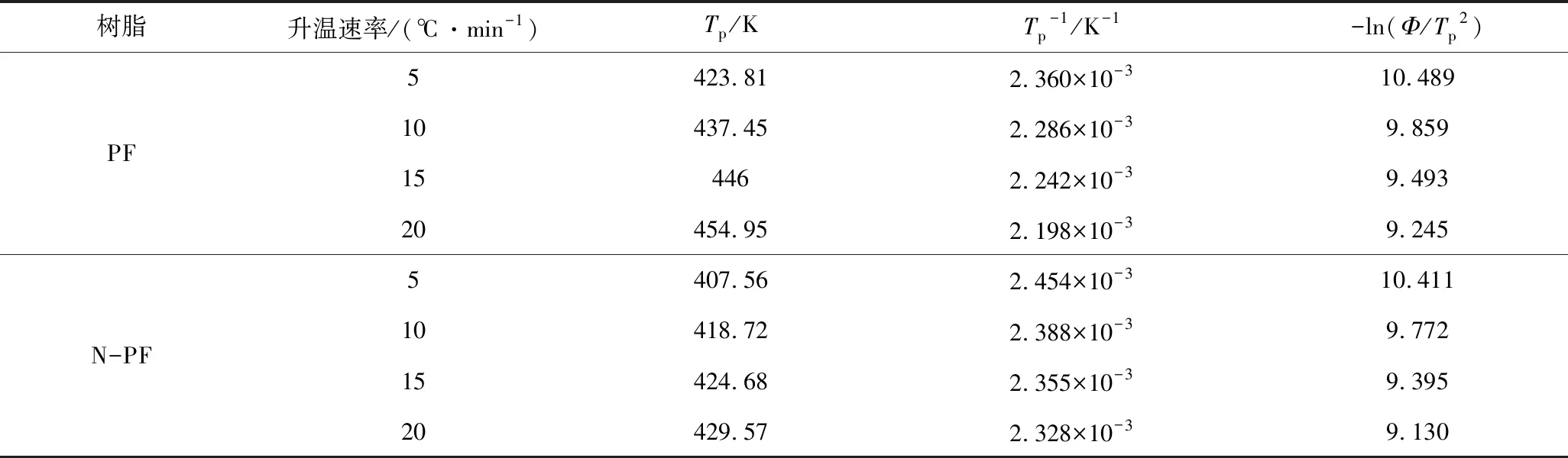
表7 Kissinger方程处理数据结果Table 7 Results using Kissinger method

表8 PF和N-PF固化动力学参数Table 8 Curing kinetic parameters for PF and N-PF
由表7可得知,PF和N-PF反应级数接近为一级反应,符合阿伦尼乌斯对一级反应活化能的表述,即活化能是分子从常态转变为过渡态所需要的平均能量,活化能越低,反应速率越快,反之反应速率越慢。由表中可以得知,N-PF的活化能为84.960 kJ/mol,远大于PF的64.863 kJ/mol,说明在固化过程中,N-PF的缩聚反应远小于PF的反应速率,再次证明了N-PF避免了集中放热,适中地拉长了反应放热曲线,使得树脂膨胀过程中的总体温度上升的程度降低,在低温固化上取得了良好的效果。
2.8 密闭空间内温度的变化分析
为了对比分析发泡过程中N-PF和PF泡沫体内部与外部温度变化,选用32 g的PF和N-PF树脂在250 mL锥形瓶中进行发泡试验,并使用热电偶监控密闭锥形瓶内的温度变化情况。在密闭空间内,PF泡沫体内最高固化温度为125 ℃,PF体系最高固化温度为41.8 ℃;N-PF泡沫体内最高固化温度为68 ℃,N-PF体系最高固化温度为32.9 ℃,发泡固化体中心温度下降46%,泡沫体边缘温度下降21%。从微观角度看,由表5可知,不同的升温速率的DSC热分析都表现出N-PF低于PF的放热焓变,证明了N-PF羟甲基数量的减少,活化能的降低,都导致整个固化体系最终放热量的降低,即泡沫体向外界散发的热量降低,对煤炭的自燃防治起着积极的作用。
3 结 论
1)确定了新型催化剂A对酚醛树脂的影响规律,优化后的N-PF的黏度为5 Pa·s、固含量为75%,更适宜发泡。通过FT-IR,13C-NMR,DSC分析和SEM图片等研究手段表明,N-PF的羟甲基指数低于PF,树脂中苯环连接结构数量与刚性均相对降低,固化放热时间延长,机械强度和力学性能略高于传统酚醛树脂。
2)通过相同条件下的发泡试验对比,发泡固化体中心温度下降46%,发泡体周围环境温度下降21%。催化剂A合成的酚醛树脂通过降低发泡固化过程的总热量和提高泡沫提热量的逃逸时间,以此来降低N-PF树脂发泡固化温度。
3)综合考虑工业应用成本和使用效果,改变酚醛树脂的催化剂能够显著提高矿用酚醛树脂发泡材料的耐储存性,在不降低发泡性能的同时降低发泡固化发泡体的温度,为进一步研究有机聚合物发泡材料在矿业领域的应用提供了新的思路。
