基于TCGA筛选结肠癌预后相关lncRNA及建立预后风险模型
2022-07-14何天曹天生
何天 曹天生
广州市花都区人民医院胃肠外科,广州 510800
结肠癌是常见的消化道肿瘤,据全球癌症统计数据,其在恶性肿瘤中发病率居第5,随着饮食结构的改变,结肠癌发病率逐渐升高[1-2]。由于结肠癌发病隐匿,疾病初期无明显临床症状,导致多数患者就诊时已处于肿瘤的中晚期,因此大多失去标准治疗的最佳时机。目前,手术切除仍是治愈的主要方式,而对有转移的晚期患者,其5 年生存率不足20%[3]。结肠癌患者预后判断主要依赖TNM 分期,血清学检查中癌胚抗原、CA19-9等生物标记物对预后评价具有重要的参考作用,但敏感性和特异性欠佳[4]。因此,临床上亟需新的生物学标志分子和有效的预后评估模型以提高对结肠癌患者诊治的准确性。
长链非编码RNA(long non-coding RNA,lncRNA)指大于200 个核苷酸的非编码RNA,一开始被认定是转录“垃圾”,不具有调节机体过程等生物学功能[5]。随着生物信息学技术与芯片技术的发展,发现lncRNA 在多种遗传生物学过程中,像转录前、后的调控和表观遗传调控等,都发挥着重要的作用[6]。研究证实当lncRNA 转录发生改变时可促进癌症的发生和转移[7]。关于lncRNA 如何影响肿瘤进程,哈佛医学院Pandolfi 课题组提出的竞争性内源RNA(competing endogenous RNA,ceRNA)假说认为,lncRNA 可通过结合微小RNA(microRNA,miRNA)来阻隔miRNA 与其他信使RNA(mRNA)作用,从而降低了miRNA 对mRNA 的抑制调控,促进相应mRNA 的表达,进而影响疾病的进程[8]。已有多种异常表达lncRNA 被证实作为ceRNA 与结肠癌进展相关,如Peng 等[9]发现lncRNA HOTAIR 在结肠癌中的表达明显升高,作为ceRNA 调节miR-34a 促进结肠癌的进展,与结肠癌的远处转移和不良预后有关。为构建1 个由lncRNA 组成的结肠癌预后风险评估模型,本研究基于癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库,获取预后相关lncRNA,构建可用于评估结肠癌患者预后的多基因风险模型,并初步探索lncRNA 参与结肠癌进程的机制。
材料与方法
1、原始数据下载及数据预处理
数据提取时间:建库至2022年3月1日。从TCGA 上下载结肠癌样本(癌和癌旁组织)转录组测序数据及临床资料。使用Rstudio 软件对获取的数据进行初步处理及筛选,根据以下标准排除部分患者:没有生存状态和生存时间信息的患者;缺乏完整lncRNA 表达数据的患者;对重复样本的患者随机去重。基因表达数据中,去除表达量较低的基因,通过从全球数据中心(Global Data Center,GDC)数据库下载人类基因注释文件(genecode.v22)进行注释。Genecode 数据库中有关于lncRNA 的种类说明,依此筛选过滤得出lncRNA的表达矩阵。
2、差异表达基因分析
依据癌旁样本挑选匹配的癌症样本构建配对样本lncRNA 表达矩阵,利用“edgeR”R 包筛选,阈值设置为:|log 2 fold change|>1.5 和P-value <0.05,获得差异表达lncRNA(differentially expressed lncRNA,DElncRNA)。根据分组绘制lncRNA 主成分分析(principal component analysis,PCA)图观察配对样本间lncRNA 的表达差异,使用“pheatmap”包绘制表达热图、“ggplot2”包绘制火山图可视化上调及下调lncRNA。
3、挑选预后相关lncRNA
构建癌组织样本DElncRNA 表达矩阵,与患者生存资料合并,使用统计学方法进一步筛选。先采用COX 回归模型单变量分析评估DElncRNA 表达水平与患者总生存期间的关系,选取明显影响生存期的lncRNA(P<0.05)。随后利用“glmnet”R 包进行Lasso 回归分析,过滤基因间的共线性因素,根据参数中的Lambda 值,选择Lambda.min 作为临界点,进行筛选。再以lncRNA 表达值的中位数作为分界值,将患者分位高低表达组,进行K-M 生存分析,挑选出表达量与生存率明显相关的lncRNA(P<0.05)。最后使用“survival”R 包的逐步回归法构建多元COX 回归模型,衡量各个lncRNA 表达情况对生存的影响程度,用风险比(hazard ratio,HR)体现,以P<0.05 作为标准,得出预后相关lncRNA[10]。
4、构建预后风险模型
根据多因素COX 回归分析中计算得出预后相关lncRNA 的回归系数(β),按照如下公式构建结肠癌预后风险评估模型[11]。β 表示相对危险度,是对模型中各个因素相对危险程度的量化。当β>0时,对应因素的取值越大,患者死亡的风险越高;β<0 时,对应因素取值越大,患者死亡的风险越低。
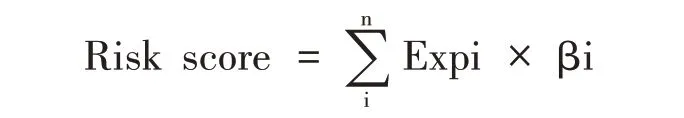
公式中Risk score(RS)为风险评分,n 为关键lnRNA 基因数量,Exp为lncRNA表达量,β为回归系数。
5、评估预后风险模型
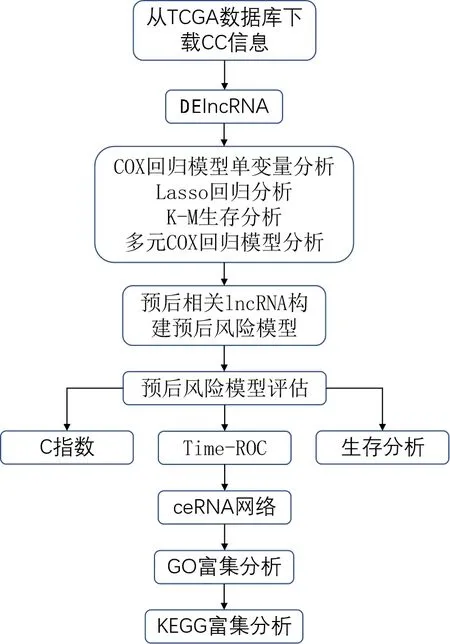
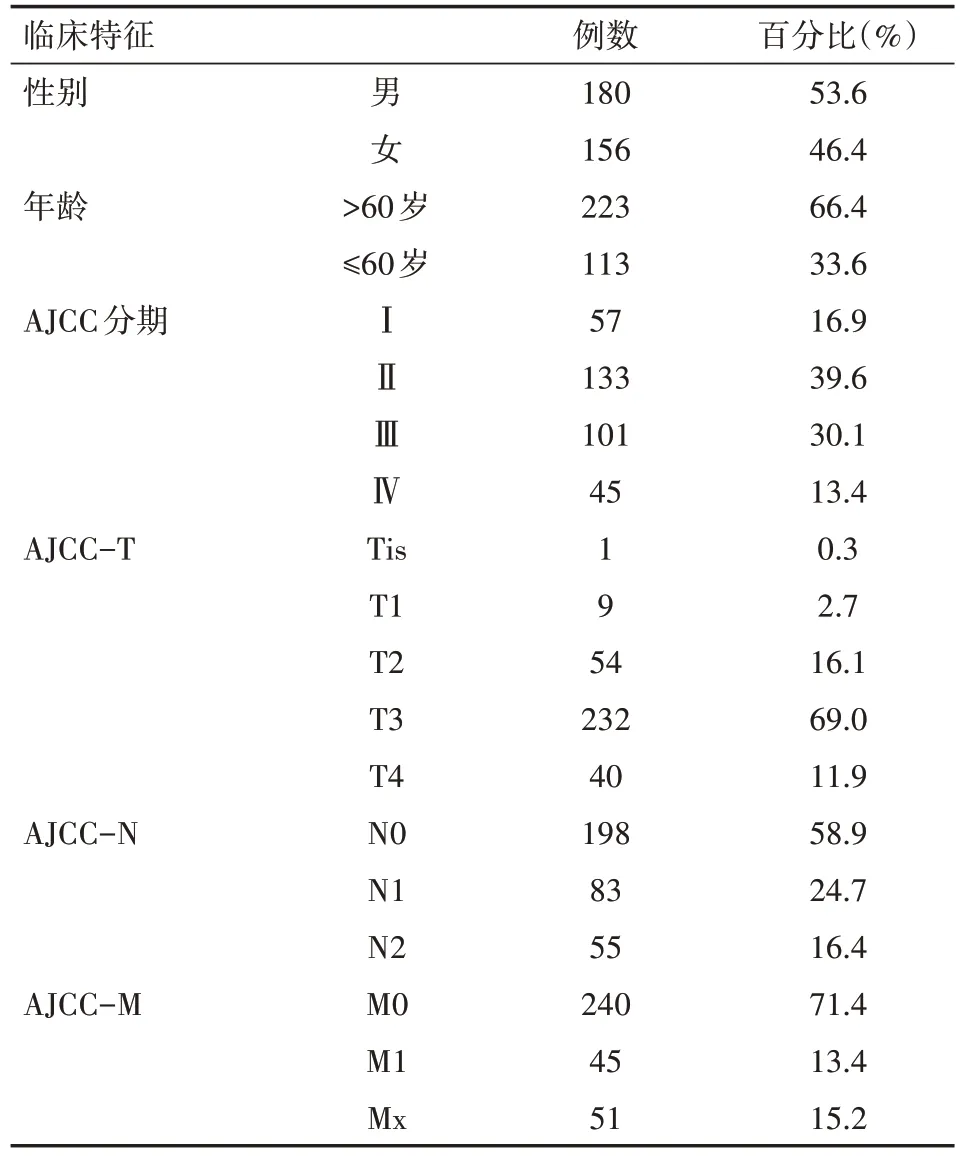
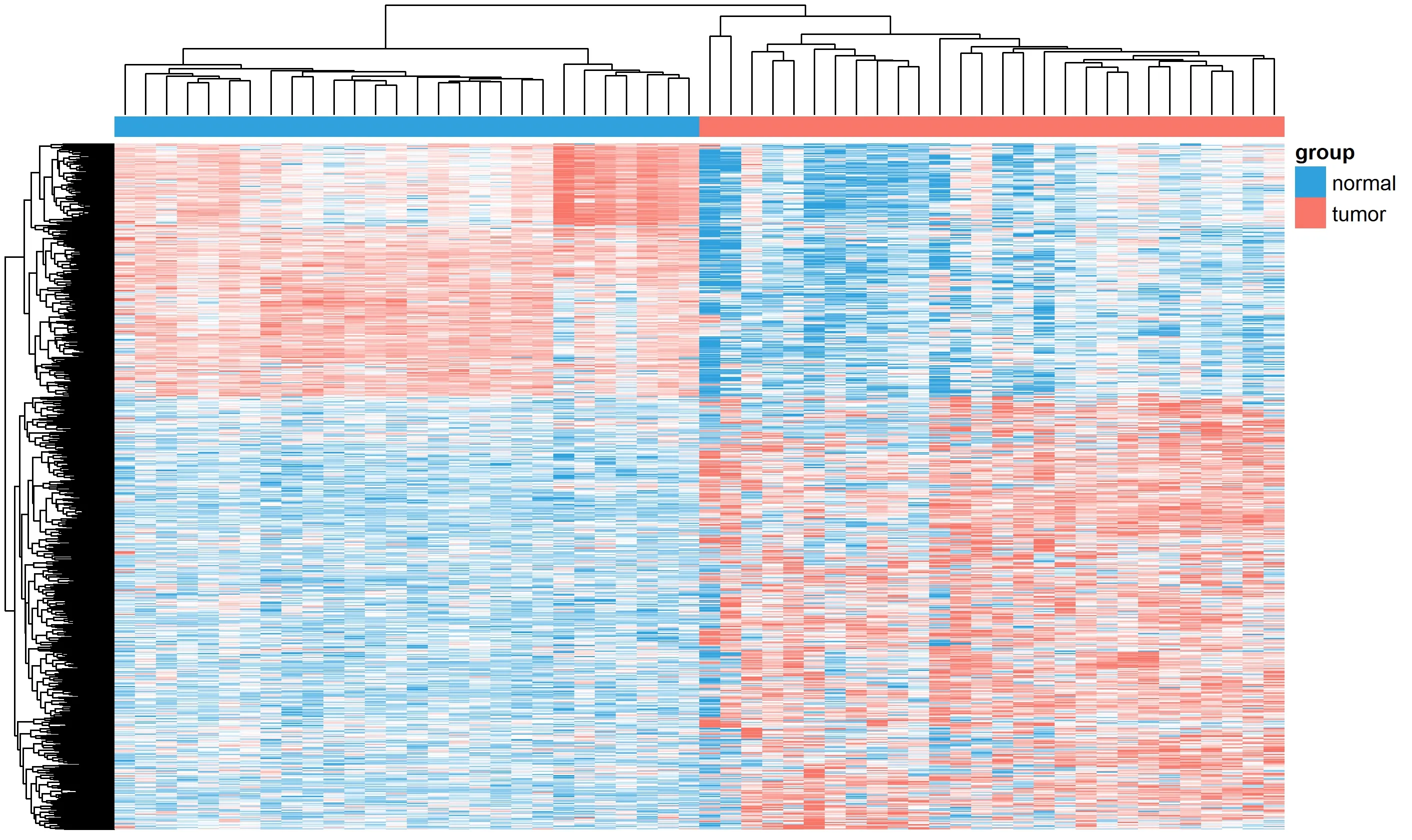
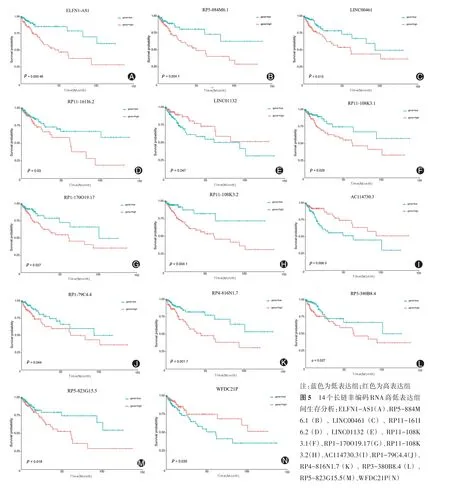
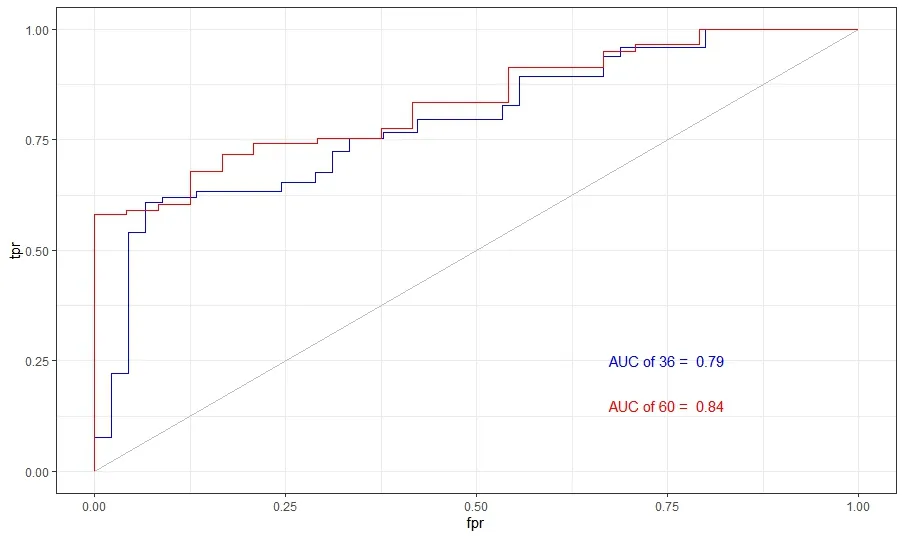
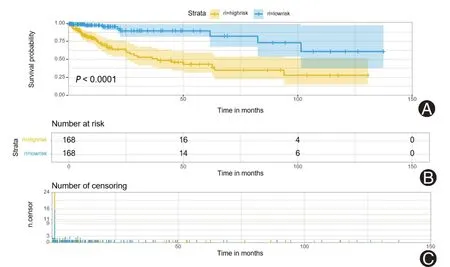
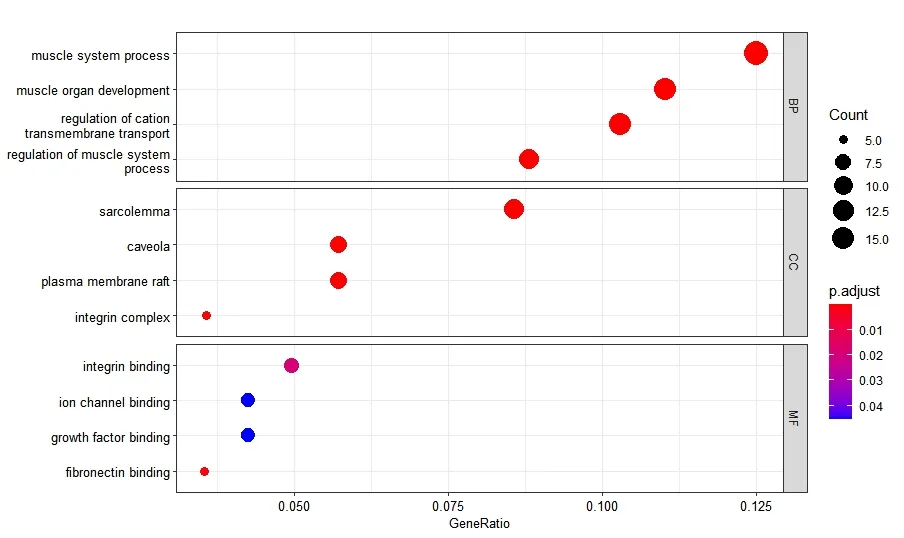
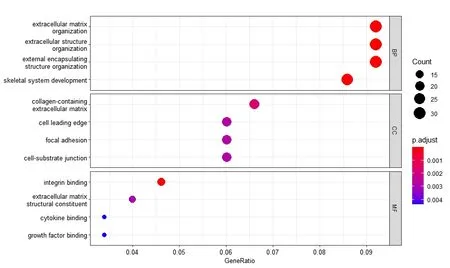
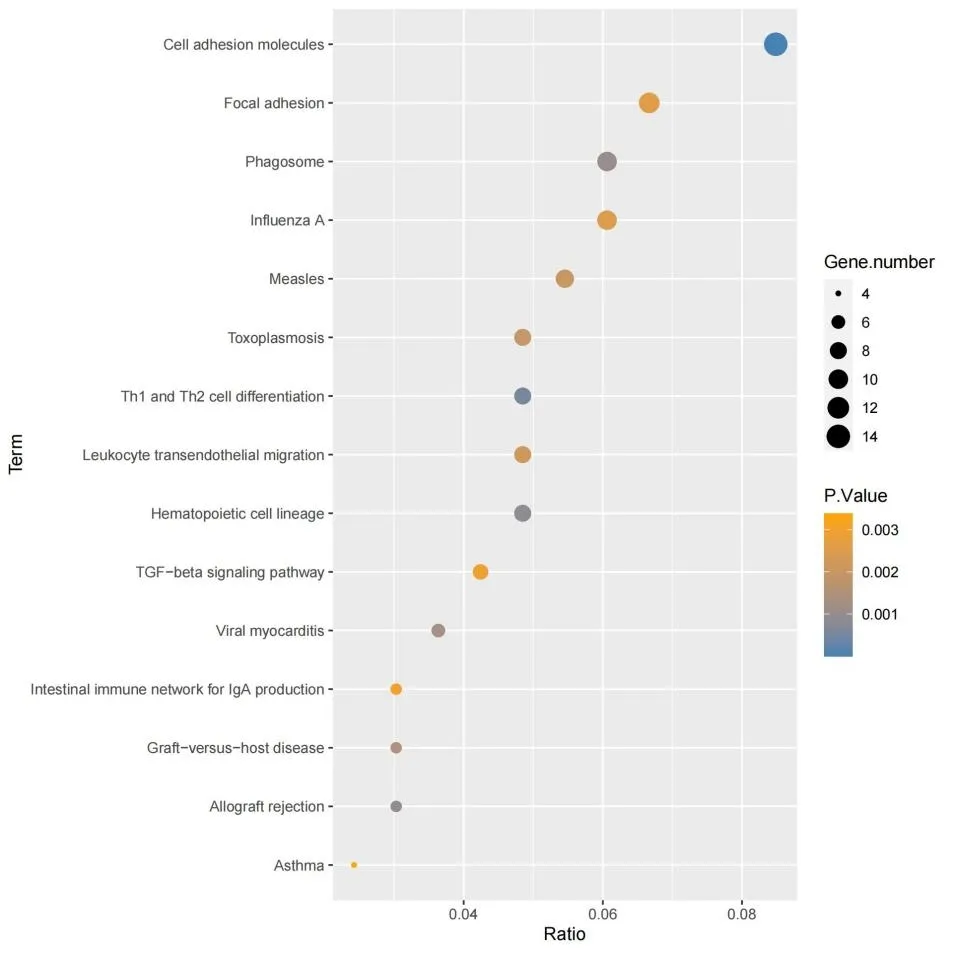
先计算模型的C 指数值,其可用来评价模型的预测能力,主要用于计算COX 回归模型中预测值与真实之间的区分度[12]。一般认为,如C指数介于0.50~0.70之间则为低准确度;在0.71~0.90 之间则视为中等准确度;高于0.90 则可看做高准确度。然后对此模型构建时间依赖的受试者工作特征曲线(receiver operating characteristic curve,ROC),并计算ROC 曲线下的面积(area under the curve,AUC),以评估模型预测患者3 年、5 年生存期的能力。AUC 和C 指数意义相似,一般认为0.85 进一步构建lncRNA-miRNA-mRNA 相互作用ceRNA网络。先利用LncBOOK 在线数据库,根据相互作用得分,选择前30个靶miRNA。然后通过数据库miRDB、TargeScan和miRTarBase 预测miRNA 下游的靶mRNA,取三者交集。在TCGA 下载结肠癌miRNA-seq 表达数据,与RNA-seq 进行整合,获得一个含有lncRNA、miRNA、mRNA 的表达数据。计算miRNA 与靶mRNA 间的皮尔逊相关系数(Pearson correlation coefficient,PCC),得出与miRNA 表达明显相关(|PCC|>0.4,P<0.05)的靶mRNA。去掉部分没有靶mRNA的miRNA,构成ceRNA 网络,使用Cytoscape 软件构图以可视化。GO 分析从生物过程(biological process,BP)、细胞组分(cellular component,CC)、分子功能(molecular function,MF)3 方面注释一系列生物学过程[14]。KEGG 富集分析可以将基因组信息与高阶的功能信息联系起来[15]。依据匹配到的mRNA,使用“ClusterProfiler”R包,进行GO、KEGG富集分析探索预后相关lncRNA 影响结肠癌进展可能的机制。整个结肠癌TCGA数据提取及分析流程见图1。 图1 结肠癌TCGA数据提取分析流程图 从TCGA 共下载514 个结肠癌样本转录组数据和452 个患者临床病理信息。依据排除标准得到364 个样本(癌旁样本28 个)和336 例患者信息,对其临床病理信息进行统计(表1)。其中,男性患者占53.6%,女性患者占46.4%;患者年龄31~90 岁,平均65.9 岁;生存或随访时间0.03~137.40个月,中位生存时间为5.62个月。 表1 336例结肠癌患者的临床病理信息 构建28 对癌与癌旁组织配对的lncRNA 表达矩阵,以|log2 FC|>1.5 和P<0.05 为标准,共获得868 个DElncRNA,绘制PCA 图(图2)、表达热图(图3)及火山图(图4)。其中PCA 图显示DElncRNA 在癌与癌旁组织中的表达具有明显差异;热图显示出在癌组织样本表达量增加的上调lncRNA及表达量下降的下调lncRNA;火山图显示上调lncRNA 548个、下调lncRNA 320个。 图2 配对样本长链非编码RNA表达主成分分析图 图3 配对样本长链非编码RNA表达热图 图4 长链非编码RNA火山图 构建癌组织DElncRNA 表达矩阵,先进行单因素COX回归分析,筛选出40 个lncRNA。随后进行Lasso 回归模型分析,得到34 个候选lncRNA。以lncRNA 表达值的中位数作为分界值,进行高低分组生存分析,发现其中的14 个lncRNA 表达谱与生存时间明显相关(P<0.05),绘制K-M 生存曲线(图5)。 图5 14 个长链非编码RNA 高低表达组间生存分析:ELFN1-AS1(A)、RP5-884M 6.1(B)、LINC00461(C)、RP11-161I 6.2(D)、LINC01132(E)、RP11-108K 3.1(F)、RP1-170O19.17(G)、RP11-108K 3.2(H)、AC114730.3(I)、RP1-79C4.4(J)、RP4-816N1.7(K)、RP3-380B8.4(L)、RP5-823G15.5(M)、WFDC21P(N) 最后进行多元COX 回归模型分析,得到7 个预后相关lncRNA(ELFN1-AS1、RP5-884M6.1、LINC00461、LINC01132、RP1-79C4.4、RP4-816N1.7、RP3-380B8.4)。计算得出每个lncRNA 的HR 值及95%可信区间,绘制风险森林图(图6)。其中1 个lncRNA(LINC01132)的HR<1,提示其表达量增加可降低结肠癌不良预后的风险,是患者的保护因素;6 个 lncRNA(ELFN1-AS1、RP5-884M6.1、LINC00461、RP1-79C4.4、RP4-816N1.7、RP3-380B8.4)的HR>l,提示它们表达量增加可增加结肠癌不良预后的风险,是患者的危险因素。 根据多元COX 回归模型,提取每个lncRNA 的回归系数,依据公式构建由7 个lncRNA 组成的结肠癌预后风险模型。 Risk score=0.256 8×(ELFN1-AS1 表达量)+0.289 1×(RP5-884M6.1 表达量)+0.244 6×(LINC00461 表达量)-0.424 4×(LINC01132表达量)+0.235 5×(RP1-79C4.4表达量)+0.298 1×(RP4-816N1.7表达量)+0.341 4×(RP3-380B8.4表达量) 通过计算模型C 指数值为0.82(图6),介于0.71~0.90之间,为中等准确度。构建时间依赖ROC,并计算AUC值。预测3 年、5 年结肠癌患者生存率的AUC 值分别为0.79 和0.84(图7),0.7 图7 风险模型时间依赖受试者工作特征曲线 根据模型计算出每个患者的RS 值,以中位数作为截断值分组,其中高、低风险组各有168 例,绘制风险评分曲线图、生存状态散点图和7 个lncRNA 的表达热图(图8)。从生存状态散点图中我们可以发现,高风险组中有更多患者处于死亡状态,且生存时间较短,并与RS呈正相关,证明模型预测的准确性。表达热图中,高风险组的ELFN1-AS1、RP5-884M6.1、LINC00461、RP1-79C4.4、RP4-816N1.7、RP3-380B8.4 表达量较高,而LINC01132 基因的表达量较低,与以上分析结果一致。 图8 风险评分3图联动。A:评分曲线图;B:生存状态散点图;C:lncRNA表达热图 对高、低风险组进行生存分析,绘制总生存期的K-M生存曲线(图9),显示高风险组患者的总体生存率更低,两组比较差异有统计学意义(P<0.000 1),且高风险组某一节点存活人数更少、单位时间内的死亡人数更多。以上说明此模型可较好评估结肠癌患者预后。 图9 风险评分高低组间生存分析图。A:生存曲线图;B:随访某一时间节点两组间存活例数;C:随访某一时间段死亡例数 对模型中的5 个lncRNA(ELFN1-AS1、RP5-884M6.1、LINC00461、LINC01132、RP1-79C4.4)构建ceRNA 网络。通过数据库lncBOOK 预测miRNA,通过miRDB、TargeScan 和miRTarBase 数据库预测mRNA,并根据相关性筛选构成lncRNA-miRNA-mRN 的ceRNA 网络,使用软件Cytoscape绘图。 对下调lncRNA(LINC01132)匹配到的154个mRNA及上调lncRNA(ELFN1-AS1、RP5-884M6.1、LINC00461、RP1-79C4.4)匹配到的的351 个mRNA 进行GO 富集分析。下调lncRNA 相关mRNA 涉及肌肉系统进程(muscle system process)、肌肉器官发展(muscle organ development)等生物过程;富集在肌纤维膜(sarcolemma)、小凹(Caveola)等细胞组件;参与整合蛋白绑定(integrin binding)、离子通道绑定(ion channel binding)等分子功能(图10)。上调lncRNA 相关的mRNA 涉及细胞外基质组织(extracellular matrix organization)、细胞外结构组织(extracellular structure organization)等生物过程;富集在内含胶原蛋白细胞外基质(collagen-containing extracellular matrix)、细胞重要边缘(cell leading edge)等细胞组件;参与整合蛋白绑定(integrin binding)、细胞外基质结构成分(extracellular matrix structural constituent)等分子功能(图11)。 图10 下调长链非编码RNA基因本体论(GO)富集分析图 图11 上调长链非编码RNA基因本体论(GO)富集分析图 再对lncRNA 相关mRNA 进行KEGG 富集分析,分组显示前15 条通路,发现下调lncRNA 相关基因显著富集在肌动蛋白细胞骨架的调控(regulation of actin cytoskeleton)、癌症中的蛋白聚糖(proteoglycans in cancer)、PI3K-Akt 信号通路(PI3K-Akt signaling pathway)等通路(图12);上调lncRNA相关基因显著富集在细胞粘附分子(Cell adhesion molecules)、局灶性粘连(Focal adhesion)、吞噬体(phagosome)等通路(图13)。 图12 下调长链非编码RNA京都基因与基因组大百科全书数据库(KEGG)富集分析图 图13 上调长链非编码RNA京都基因与基因组大百科全书数据库(KEGG)富集分析图 lncRNA 在细胞增殖分化、血管再生和细胞活力等方面发挥了重要的作用,当其表达发生改变时可影响肿瘤的发生和转移[16]。随着生物学研究方法不断进步,越来越多lncRNA 被深入研究,通过对其进行整合分析,构建了多种癌症不同的预后风险模型,对肿瘤预后判断提供了重要帮助[17-18]。大量研究表明,lncRNA 参与了结直肠癌的进展,并可能成为诊断、治疗及预后判断的潜在生物标志物[19]。 研究中,我们使用TCGA 数据库结肠癌患者的转录组数据进行分析并构建预后风险模型。分析方法上,我们在COX回归分析的基础上进行Lasso回归分析,能有效过滤其中的共线性因素,构建了一个更为简练而准确的模型。并对每个lncRNA 进行单独的生存分析,保证了模型中每个lncRNA 均对结肠癌患者生存率具有显著影响。在模型准确性评估方面,通过多种指标进行评估,包括C 指数值、ROC及AUC值、RS值可视化和高、低风险组生存分析,均表明模型具有较好的结肠癌预后预测能力。本研究筛选出7 个lncRNA 构成的结肠癌预后风险模型,与单一的生物标志物相比,多个综合的生物标志物系统可以提高容错性和预测的准确性。目前此模型在国内外的研究中未有报道,提示其可为结肠癌的预后判断提供新的参考,及为结肠癌的分子机制研究提供新的方向。 模型中的7 个lncRNA 均被Ensembl 数据库所注解,关于它们在肿瘤中的作用已有不同程度的认识。关于下调lncRNA(LINC01132),研究发现其在胶质母细胞瘤维持缺氧诱导因子的高活性和肿瘤细胞迁移率[20];有研究通过生物分析发现其在卵巢癌中低表达,与患者的生存率明显相关,可用于评估卵巢癌患者预后,与本研究结果相符[21]。对于ELFN1-AS1则有较多的报道,如Lei等[22]发 现ELFN1-AS1 通过调节MIR-4644/TRIM44 轴加速了结直肠癌的增殖和迁移。LINC00461 是一个明星lncRNA,研究发现LINC00461 在多种癌症中高表达,通过不同的途径影响着肿瘤的发生、进展,其中包括结直肠癌[23-24]。Fu等[25]通过TCGA 数据分析发现RP4-816N1.7 与结肠腺癌预后密切相关。模型中的4个lncRNA(LINC01132、ELFN1-AS1、LINC00461、RP4-816N1.7)均有相关的研究报道,大多与结肠癌有关,从侧面验证本研究预后模型的准确性。然而根据PubMed数据库的搜索,没有发现模型中其余3个lncRNA(RP5-884M6.1、RP3-380B8.4、RP1-79C4.4)研究报道,其在肿瘤,特别是结肠癌中的作用机制有待发掘。 为了探究模型中的lncRNA 潜在的生物学功能及如何影响结肠癌进程,本研究构建ceRNA 网络,通过GO 分析发现,lncRNA 相关mRNA 基因主要富集在细胞膜、细胞质等细胞组件,参与整合蛋白绑定、离子通道绑定、细胞外基质结构成分等分子功能,涉及肌肉系统进程、肌肉器官发展、细胞外基质组织、细胞外结构组织等生物过程。KEGG 富集分析提示下调lncRNA 可能是通过肌动蛋白细胞骨架的调控、癌症中蛋白聚糖、PI3K-Akt 信号通路等抑制结肠癌进展,上调lncRNA 可能是通过细胞粘附分子、局灶性粘连、吞噬体等通路促进结肠癌进展。 虽然我们构建了一个有效的结肠癌预后风险模型,但也存在着一些不足。首先,从TCGA 中提取的结肠癌样本量不够大;其次,模型的预后价值评估不是十分令人满意;再者,lncRNA、miRNA 和mRNA 间的相关程度及lncRNA 如何影响结肠癌进程主要通过在线数据库进行预测,需要进一步的实验研究。 综上所述,本研究对TCGA 中结肠癌转录组数据进行整理、分析,筛选出7 个预后相关lncRNA,构建预后风险模型,通过评估证明其具有较好预测患者生存预后的准确性,同时每个lncRNA 是潜在单独的预后生物标志物,对结肠癌患者临床预后评估具有一定的参考价值。通过GO、KEGG富集分析对模型中lncRNA 影响结肠癌机制进行初步探索。在临床上,结合结肠癌患者的lncRNA 分子水平的风险评分,可筛选出高风险患者,制定个体化的治疗方案,有利于患者病情的预后评估和综合诊治。6、构 建ceRNA 网络及基因本体论(Gene Ontology,GO)、京都基因与基因组大百科全书数据库(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析
结果
1、TCGA中结肠癌患者临床病理信息
2、配对样本差异分析结果


3、结肠癌预后关键lncRNA的筛选
4、预后风险模型的构建

5、预后风险模型的评估

6、ceRNA网络构建及GO、KEGG富集分析

讨论
