黄体生成素对卵母细胞减数分裂恢复与排卵过程的分子机制研究进展
2022-07-13林乐丰范冰峰许保增
林乐丰,范冰峰,许保增
(特种经济动物分子生物学重点实验室,中国农业科学院特产研究所,吉林长春 130112)
黄体生成素(Luteinizing Hormone,LH)是由下丘脑促性腺激素释放激素刺激垂体前叶分泌的一种异源二聚体糖蛋白激素,它在结构和功能上与人绒毛膜促性腺激素相关,在卵泡生长发育和排卵过程中起着重要作用,LH 在所有物种中(包括人类)都有分布,在一个生殖周期中,LH 激增会使已分裂停滞的卵母细胞恢复减数分裂,继续其细胞周期。LH 激增前,减数分裂阻滞由卵母细胞内高水平环磷酸腺苷(Cyclic Adenosine Monophosphate,cAMP)维持,壁颗粒细胞表达的C 型钠肽与卵丘细胞中钠肽受体2(Natriuretic Peptide Receptor 2,NPR2)结合产生环磷酸鸟苷(Cyclic Guanosine Monophosphate,cGMP),通过缝隙连接运送到卵母细胞阻止cAMP 降解。LH 激增后,高水平的LH 作用于黄体生成素受体激活G 蛋白偶联受体活性,使壁颗粒细胞中cAMP 水平升高。高水平cAMP 激活丝裂原活化蛋白激酶、表皮细胞生长因子(Epidermal Growth Factor,EGF)等信号通路,使其参与微管的装配与纺锤体的组装,以及第一、第二极体排出。本文从LH 对下游不同信号通路的激活和抑制及调节卵母细胞成熟和排卵的机制进行综述,为获得高质量卵母细胞,提高雌性良种家畜扩繁效率提供理论基础。
1 卵母细胞第1 次减数分裂阻滞的调控机制
哺乳动物体内卵母细胞在胎儿期就已进行分化,并停滞在第1 次减数分裂前期的末期,即双线期。cAMP 是存在于卵母细胞内的第2 信使,高水平的cAMP 对哺乳动物卵母细胞减数分裂恢复具有抑制作用。细胞周期蛋白依赖性激酶和细胞周期蛋白B 结合,形成成熟促进因子对减数分裂周期具有调控作用,cAMP 激活蛋白激酶A(Protein Kinases A,PKA)信号通路从而抑制周期蛋白激酶活性,阻止减数分裂恢复。卵母细胞膜上有许多G 蛋白偶联受体,其中GPR3 和GPR12 对减数分裂的阻滞起作用,并在小鼠、大鼠和非洲爪蟾的实验中得到证实,它可将胞外信号传递给胞内Gs 蛋白,使腺苷酸环化酶活化,使卵母细胞产生cAMP。卵母细胞中存在的磷酸二酯酶3 在有活性的情况下会促进cAMP 降解,恢复减数分裂,细胞中cGMP 的存在能够抑制磷酸二酯酶3 活性。颗粒细胞表达的C 型钠肽作用于卵丘细胞中NPR2 生成cGMP,通过缝隙连接蛋白37 进入卵母细胞。卵母细胞自身并不表达NPR2,也不能自行产生cGMP。卵母细胞中高水平cGMP 是阻止细胞减数分裂恢复的主要原因。颗粒细胞通过缝隙连接使卵母细胞停滞在减数第一次分裂,这种调控系统已在小鼠等哺乳动物身上得到验证。小鼠卵母细胞减数分裂的信号通路如图1 所示。
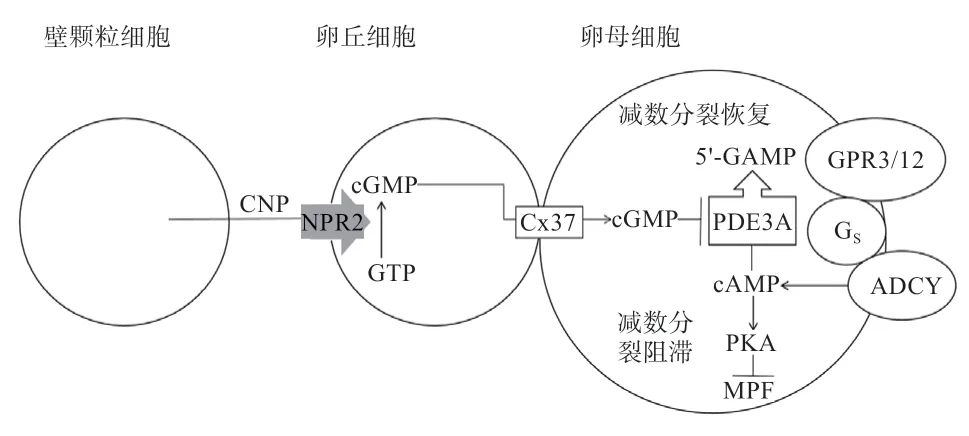
图1 小鼠卵母细胞减数分裂的信号通路(图片参考Zhang 等[8])
2 LH 信号恢复卵母细胞第一次减数分裂的机制
2.1 LH 结合受体激活EGF 和MAPK3/1 下游信号通路减数分裂恢复过程的信号通路如图2 所示。LH 激增并与颗粒细胞上受体结合激活G 蛋白偶联受体,提高细胞中cAMP 水平。cAMP 激增是后续众多信号通路的关键,高水平cAMP 可以增加EGF 表达,使EGF 与受体EGFR 结合,EGFR 激活是缝隙连接关闭和卵母细胞cGMP 降低的部分原因。大鼠排卵前LH 峰会诱导表调节蛋白和双调节蛋白表达,它们都属于EGF家族成员。这些EGF 样因子激活了丝裂原活化蛋白激酶3/1(Mitogen-activated Protein Kinases 3 and 1,MAPK3/1),也被称为细胞外信号激酶1/2(Extracellular Signal-regulated Kinases 1 and 2,ERK1/2)通路。MAPK 是一种丝氨酸/苏氨酸蛋白激酶,ERK1/2 是其中的一个亚族,在减数分裂恢复过程中起着重要作用。卵母细胞体外实验证明,ERK1/2 活性对自发的减数分裂恢复并不是必需的,但对于卵丘细胞是必要的,即使用外源激素处理ERK1/2小鼠,也没有卵母细胞成熟和生发泡破裂。ERK1/2 激活促进前列腺素内过氧化物合成酶2 在卵泡液中合成前列腺素E2。缺乏过氧化物合成酶2 或前列腺素受体2 的小鼠排卵数少且卵母细胞完全丧失受精能力,表明前列腺素E2 在排卵过程中起到重要作用。此外,前列腺素E2 可以通过诱导cAMP/PKA 和MAPK3/1 通路合成表调节蛋白和双调蛋白来模拟LH 的作用。上述结果表明EGF 样因子和前列腺素E2 之间存在着正反馈作用。

图2 减数分裂恢复过程的信号通路(图片参考Richards 等[9])
最近有研究根据基因芯片数据和定量逆转录结果,证明许多与排卵相关的基因在ERK1/2的卵巢中不表达。因此,LH 最初表达表调节蛋白和双调节蛋白在很大程度上不依赖于ERK1/2 通路。然而表调节蛋白、双调节蛋白的后续表达可能依赖于ERK1/2 对前列腺素内过氧化物合成酶2 的作用,使前列腺素E2 高表达。肿瘤坏死因子转化酶被证明是小鼠胚胎细胞中表调节蛋白和双调节蛋白的主要释放酶,cAMP 依赖途径激活颗粒细胞和卵丘细胞表达肿瘤坏死因子转化酶,释放EGF 样因子激活EGF-MAPK3/1 通路。此外,增强子结合蛋白-b 缺失小鼠卵巢表型与Erk1/2表型相似。只有在增强子结合蛋白-b 表达的细胞中,双调蛋白才能诱导ERK1/2 依赖的靶基因()表达。因此,增强子结合蛋白-b 是ERK1/2 直接底物,两者共同构成了颗粒细胞重要信号网络。
2.2 EGF 和MAPK3/1 信号通路降低卵母细胞中cGMP含量 利钠肽前体C(NPPC)作用于卵丘细胞上钠肽受体(NPR2)提高卵丘细胞中cGMP 水平,阻止减数分裂恢复,因此下调NPPC 和NPR2 信号通路在减数分裂恢复过程中起着重要作用。相应的,NPPC 或NPR2 缺失或突变使小鼠恢复减数分裂。有实验证明,用双调蛋白作用于小鼠卵泡培养液,其中NPPC 的受抑制程度与使用LH 的结果相同,EGFR 激酶抑制剂AG1478 可以消除双调蛋白对NPPC 的抑制作用,说明LH-双调蛋白-EGFR 信号通路的激活可以下调NPPC表达。同样地,在小鼠体外培养卵丘卵母复合体实验中,EGFR 信号激活使NPR2 表达水平显著下降,但是减数分裂过程仍然受阻,可能是由于细胞中NPR2 半衰期较长,卵丘细胞中NPR2 水平并未降低。EGF可以提高卵丘细胞中钙水平,使NPR2 失活,降低与NPPC 结合力,减少相互作用,从而恢复减数分裂。NPR2 失活还与其去磷酸化有关。用抑制丝氨酸-苏氨酸磷酸化酶中的PPP 家族试剂斑泻素 100 μmol/L 或冈田酸 10 μmol/L 预先孵育卵泡1h,可阻止NPR2 活性降低。这些结果表明PPP 家族磷酸酶活性是NPR2 去磷酸化所必需的,而LH 诱导NPR2 活性降低也需要去磷酸化。LH 介导雌二醇-雌二醇受体信号通路同样可以降低NPPC 和NPR2 表达,雌二醇受体包括(ER和ER)。在对和启动子进行染色质免疫共沉淀的实验中,小鼠载体和载体的共转染显著增强了荧光素酶活性,表明ER、ER可以加强和基因的转录。且在ER、ER过表达的细胞中,雌二醇显著促进NPPC/NPR2 表达,因此雌二醇-雌二醇受体通路可以增加颗粒细胞中NPPC 和NPR2 含量,阻止减数分裂的恢复。在人超数排卵实验中,卵丘细胞中LH 受体含量与血清中雌二醇呈负相关,LH 激增后,LH 受体在卵丘细胞中表达,雌二醇水平下降,NPPC 和NPR2 含量降低,减数分裂恢复。原因是垂体分泌卵泡刺激素(FSH)作用于受体后增加芳香化酶表达,而LH 激增会降低这种作用,使产生雌二醇的颗粒细胞转化为产生黄体细胞。
在大鼠和小鼠颗粒细胞中cGMP 水解由cGMP 结合磷酸二酯酶(PDEs)活性决定,主要是PDE1A 和PDE5。PDE5 的N 端有2 个与cGMP 结合的高度同源结构域,为GAF A 和GAF B。cGMP 与GAF A 结合改变PDE5 构象,直接激活PDE5 水解活性,而不需要使其磷酸化。另一方面,LH 信号在传导过程中增加了PKA 含量,诱导PDE5 磷酸化,激活其活性,从而进一步降低颗粒细胞中cGMP 含量。用腺苷酸磷酸化酶激活剂Forskolin 处理的大鼠卵泡PDE5 活性增加,与LH的反应结果相同,证明PKA 和PKG 磷酸化PDE5 上丝氨酸位点是LH 诱导PDE5 活性增加的原因。此外,PDE5 活性增加与NPR2 去磷酸化导致的cGMP 下降是同时发生。
哺乳动物的缝隙连接通道由连接蛋白基因家族蛋白组成,这些通道允许离子、氨基酸、核苷酸和二级信使(如Ca、葡萄糖、cAMP、cGMP)在细胞之间扩散。其中连接蛋白43(Connexin 43,Cx43)形成壁颗粒细胞间缝隙连接,维持成熟卵母细胞减数分裂停滞的调节信号cGMP 通过缝隙连接从壁颗粒细胞扩散到卵母细胞中。在减数分裂恢复过程中,LH 通过磷酸化降低其活性,使缝隙连接关闭,减少cAMP 扩散水平以重启细胞周期,这一过程是由MAPK 家族介导。LH 作用于受体后,刺激EGF、TPA 等物质产生,引起Cx43 上特定氨基酸位点顺序磷酸化,缩短其半衰期并使缝隙连接关闭。EGF 诱导AKT 磷酸化Cx43 上S373 和S369位点,诱导抑制Cx43:ZO-1 间相互作用,减少传入缝隙连接通道的质膜;MAPK 可以使Cx43 上S255、S279、S282 位点磷酸化,使缝隙连接关闭;最后PKC使S368 位点磷酸化。这些残基磷酸化已被证明会影响缝隙连接通道门控特性和/或缝隙连接组装减少,活化的src 也可以下调缝隙连接。LH 作用于受体后并不会降低Cx37 活性,卵母细胞和卵丘细胞之间缝隙连接没有受到显著影响。这可能是因为卵母细胞减数分裂过程中需要卵丘细胞提供氨基酸、胆固醇等物质,并将细胞中cGMP 运输到卵丘细胞中。
3 LH 调控卵母细胞排卵的分子机制
3.1 LH 激活孕激素受体的表达 排卵是由LH 激增启动,它直接作用于卵泡膜和壁层颗粒细胞,诱导卵母细胞胞质和核成熟的最后阶段,随后卵泡破裂、黄体形成。排卵前卵母细胞并不表达LH 受体,尽管有报道称卵母细胞周围的卵丘细胞含有该受体,但是LH 信号强度明显低于颗粒细胞。因此,LH 信号要到达卵母细胞,需要先产生第二信使cAMP 并通过缝隙连接将cAMP 传到卵泡中。LH 与壁颗粒上的受体相互作用,激活腺苷酸环化酶,使卵泡内第二信使cAMP 含量显著上升。cAMP 升高导致PKA 激活,进而激活cAMP 反应元件结合蛋白,PKA 信号通路的激活也被认为是排卵前卵泡介导LH 作用的主要途径。排卵前,卵泡表面上皮细胞开始分裂,结缔组织降解,在卵泡其他部位,颗粒细胞和囊膜细胞开始黄体化过程,颗粒细胞开始增大,并积累脂质形成小滴,为类固醇激素合成提供胆固醇。排卵前,卵泡合成孕酮,在绵羊的体内实验中,用异沙唑抑制孕酮合成,排卵过程会受到影响,此外,抗孕酮血清降低了用促性腺激素处理的未成熟大鼠排卵率,说明孕酮在排卵过程中起着不可缺少的作用。在人和猪的实验中,LH、FSH 和Forskolin 均可使孕激素受体(PGR)水平升高。孕酮受体即PGR 是一种核受体转录因子,在排卵前卵泡颗粒细胞中表达,而卵丘细胞和膜细胞几乎不表达。基因同时编码PGR-a和PGR-b,当雌性小鼠双基因敲除后,小鼠不育,卵巢有巨大的有腔卵泡,却无法启动排卵,表明PGR在排卵过程中起关键作用。虽然啮齿类动物和灵长类动物在卵巢生理(单个排卵卵泡与多个排卵卵泡)和卵泡PGR 蛋白表达方面存在差异,但孕酮在灵长类卵巢中排卵也是通过PGR 介导。在恒河猴Pgr 敲除实验中,将腺病毒载体在排卵前22 h 注射卵泡中,PGR mRNA 被转导为PGR siRNA,注射腺病毒载体卵泡中PGR mRNA 和蛋白表达受到抑制,体外颗粒细胞和体内排卵周卵泡功能丧失,且没有排卵痕迹,而对照组排卵不受到影响。
3.2 孕激素受体激活下游信号通路 内皮素-2 是一种有效的血管活性分子,在卵泡颗粒细胞中瞬时表达,这种表达在PGR 缺失的小鼠卵巢中不复存在。Palanisamy等分别使用内皮素-2 受体ETR-A 和ETR-B 的拮抗剂,发现用ETR-B 拮抗剂处理过的小鼠释放卵母细胞数量显著减少,ETR-B 在排卵前卵泡的壁细胞、卵丘颗粒细胞以及内膜内缘毛细血管中有较高表达。此外阻断内皮素-2 通路显著降低了cGMP 依赖的蛋白激酶II表达水平,说明蛋白激酶II 受颗粒细胞中内皮素-2 信号的调控。内皮素-2 通过自分泌作用于壁细胞中的ETR-B 产生蛋白激酶II,也可以通过旁分泌刺激内皮型一氧化氮合酶生成一氧化氮作用于卵丘细胞,诱导这些细胞合成蛋白激酶II。Richards 等认为蛋白激酶II 是合成急性调节蛋白的关键分子,蛋白激酶II 与排卵过程中某些功能相关。过氧化物酶体增殖物激活受体(PPAR)是排卵过程中PGR 调节的下游靶点,参与脂肪细胞分化、调节脂质和葡萄糖稳态,以及控制炎症反应。在排卵过程中,该受体基因条件性丢失虽然不会影响卵泡发育,但会阻止卵泡破裂。PPAR是一种配体诱导的转录因子,它被环氧合酶的脂肪酸代谢产物所激活,激活的PPAR通过增加内皮素-2、蛋白激酶II 和白细胞介素-6 表达介导PGR 功能,不受PPAR调节的分子如ADAMTS-1 可能也参与了排卵过程(图3)。白细胞介素-6 mRNA 可以在小鼠颗粒细胞中检测到,它是一种多功能细胞因子,也是炎症介质,具有广泛的生物活性,如增加血管通透性、参与血管生成,可能在排卵和黄体形成过程中起作用,白细胞介素-6 也是大鼠T-激肽原基因的诱导剂,在排卵过程中增加白细胞介素-6 水平,对调节蛋白水解位置和速率起着重要的作用。ADAMTS-1 是一种由PGR 诱导的卵泡细胞外金属蛋白酶,通过介导浸润血管的基质屏障降解促进排卵卵泡中卵泡结构重塑,在卵丘卵母复合体基质形成和分解代谢中起到关键作用。在ADAMTS-1的小鼠中,排卵率降低了77%,排卵卵母细胞的受精率下降63%。ADAM8 是一种金属蛋白酶,在排卵周卵泡颗粒细胞和卵丘细胞中均有表达,ADAM8 可以改变细胞外基质影响排卵过程,ADAM8 是PGR 潜在靶点,在PGR 缺失型小鼠中表达量显著下降。此外,SNAP25是可溶性N-乙基马来酰亚胺敏感因子粘附蛋白受体的重要组成因子,在囊泡胞吐过程中对细胞膜融合起重要作用,传递PGR 信号,也可以对细胞中白细胞介素-6水平进行调控。

图3 PGR 调控排卵的信号通路(参考Kim 等[45])
4 小结
LH 在卵母细胞减数分裂恢复和排卵过程中起着重要作用。LH 激增后,激活MAPK 和EGF 信号通路,使颗粒细胞中C 型钠肽与分泌在卵丘细胞NPR2 活性降低,颗粒细胞中cGMP 水解活性增加以及缝隙连接关闭,恢复卵母细胞减数分裂。LH 通过诱导PGR、EGF 表达并作用于下游信号调节分子ADAM8 和Snap25 等,增加蛋白水解酶和炎症因子活性使卵泡膜破裂,卵母细胞排出。然而对于LH 调控卵母细胞成熟和排卵仍有问题值得去探索,如现有研究都是集中在小鼠上,在大型哺乳动物及养殖生产中,现有结论是否仍旧适用有待进一步验证。了解LH 在这些过程中分子机制将为建立更加高效的卵母细胞体外成熟体系、提高体外成熟卵母细胞质量提供理论参考,也为提高雌性家畜扩繁效率、扩大畜牧养殖规模提供了理论基础。
