蛋白多肽类降糖药物口服递送载体的研究现状与临床应用进展
2022-07-01杨甜甜王奥华俞淼荣甘勇
杨甜甜 ,王奥华 ,俞淼荣 ,甘勇 ,
(1. 中国科学院上海药物研究所,新药研究国家重点实验室,上海 201203;2. 中国科学院大学,北京 100049;3. 中国食品药品检定研究院,国家药品监督管理局药用辅料质量研究与评价重点实验室,北京 100050)
糖尿病是21世纪全球最流行的慢性疾病之一。国际糖尿病联盟(International Diabetes Federation,IDF)发布的最新《全球糖尿病地图(第10版)》显示,截至2021年全球约有5.37亿成人患有糖尿病(患病率达10.5%),平均每5秒就有1人死于糖尿病。糖尿病的治疗给社会和家庭都造成了巨大的经济负担,我国的糖尿病相关卫生支出位居全球第2,共计1 650亿美元[1]。糖尿病根据发病的病因不同,可划分为1型糖尿病和2型糖尿病。其中1型糖尿病主要因胰岛β细胞受到细胞介导的自身免疫性破坏,无法合成和分泌胰岛素引起。因此,针对1型糖尿病,其他降糖药无法起效,只能终身注射胰岛素以维持血糖水平[2]。2型糖尿病则主要表现为相对胰岛素缺乏或胰岛素抵抗,患者体内的肌肉以及肝脏等器官对于胰岛素的敏感性不够,致使血糖不能被有效利用而维持在较高水平[3]。随着病程的不断延长,2型糖尿病患者的胰岛β细胞功能会逐渐衰退,导致胰岛素分泌不足,因此胰岛素是2型糖尿病三线和四线治疗中最重要的部分。此外,胰高血糖素样肽-1(glucagon-like peptide-1,GLP-1)类似物也常用于治疗2型糖尿病,可促进胰岛素的合成和分泌,抑制胰岛β细胞凋亡,并减少胰高血糖素的分泌,从而降低血糖[4-5]。近年来已上市的GLP-1类似物,如艾塞那肽、利拉鲁肽、杜拉鲁肽以及索马鲁肽等都显示了较好的临床治疗效果,成为糖尿病治疗的新选择[6]。
口服给药是最简单、常用的给药途径,也是胰岛素及GLP-1类似物等药物最理想的给药方式。然而,体内复杂的生理屏障严重阻碍了药物的口服吸收,导致其生物利用度较低。近年来,多种口服递送载体被开发出来,有望解决上述问题,进一步提高药物疗效。本文总结了体内限制药物口服递送的共性屏障,重点阐述蛋白多肽类降糖药物口服递送面临的关键屏障;并在此基础上对近年来已报道的相关口服递送载体的设计策略以及临床研究进展进行综述。
1 口服蛋白多肽类降糖药物的优势
胰岛素与GLP-1类似物同属于蛋白多肽类降糖药物,目前临床上的给药方式仍以皮下注射为主。但是,长期频繁的注射会给患者带来极大的痛苦和不便,同时还可能导致低血糖、水肿、体质量增加以及皮肤感染等不良反应。与之相比,口服给药则是一种更安全有效的非侵入式治疗策略,具有良好的患者顺应性[7]。
口服胰岛素递送还具有其他给药途径无法比拟的优势,即可模拟内源性胰岛素的分泌。在正常生理状态下,胰岛素经胰岛β细胞分泌后,首先经肝门静脉被运送至肝脏。在肝脏的首过作用下,约有50% ~ 80%的胰岛素被代谢,之后剩余的胰岛素再进入外周循环,因此就形成了肝脏-外周的胰岛素浓度梯度[8]。据计算,肝脏处的胰岛素浓度约为外周组织的2 ~ 4倍[9]。同样地,口服胰岛素经小肠吸收后由肠壁毛细血管汇入肝门静脉并被运送至肝脏,因此肝脏可暴露在较高的胰岛素浓度下,外周组织则维持相对较低的胰岛素水平,避免了由于外周高胰岛素血症引起的低血糖等不良反应。同时,肝脏对于胰岛素高度敏感,高胰岛素暴露量不仅可以促进肝脏对于葡萄糖的摄取和利用,还可有效抑制肝糖原的分解,从而更好地调节血糖水平[10]。此外,有研究发现口服胰岛素还具有免疫调节的作用。通过口服低剂量的胰岛素可诱导T细胞的抗原反应性,从而产生免疫耐受,保护胰岛β细胞免受自身免疫性破坏,有助于预防或延缓1型糖尿病的发生与发展[11-13]。
2 蛋白多肽类降糖药物口服递送面临的挑战
胃肠道的pH和酶通常被认为是限制蛋白多肽类药物口服吸收的主要因素[14]。故多采用药物载体如肠溶胶囊或纳米粒、脂质体、微球等微纳米载体将药物包载在内部,提高药物在胃肠道的稳定性[6]。随着研究的不断深入,研究人员发现体内还存在着多重屏障制约药物载体的口服吸收以及后续入血发挥作用(见图1)。一方面,由小肠黏液和上皮细胞构成的物理化学屏障以及肠道内的蛋白冠冕作用等,它们极大地阻碍了药物载体的转运,成为限制药物口服递送的共性屏障。另一方面,针对胰岛素等蛋白多肽类降糖药物的口服递送,药物载体的胞内转运、基底侧出胞过程以及在肝脏的处置作用同样至关重要,它们在一定程度上影响了蛋白多肽类降糖药物作用的发挥,成为导致该类药物口服生物利用度低下的特殊屏障。

图1 体内限制蛋白多肽类降糖药物口服递送的屏障示意图Figure 1 Schematic illustration of the barriers restricting oral delivery of hypoglycemic proteins and peptides in vivo
2.1 口服药物载体面临的共性屏障
小肠的内表面积大,转运时间长,有利于药物载体的吸收。但是,小肠内的黏液、肠上皮细胞屏障以及蛋白冠冕作用等,共同限制了药物载体的口服吸收效率,导致药物生物利用度低[15]。因此,药物载体在到达靶部位发挥作用前,首先需要克服小肠内制约载体口服递药的共性屏障。
2.1.1 黏液屏障小肠黏液是一种具有黏弹性的凝胶型网状结构,孔隙大小介于100 ~ 200 nm[16]。黏液层可以保护肠上皮细胞不直接暴露于肠腔环境,但也限制了口服药物载体到达肠上皮细胞。黏蛋白是黏液中最主要的结构和功能成分[17],在肠道的pH条件下黏蛋白带负电,且表面分布着高密度的疏水区域,因此可与表面带正电且具有疏水性的药物载体发生相互作用,并将其固定在黏液中[18]。小肠黏液的更新速度较快,每隔4 ~ 5 h更新1次,有助于快速清除黏附的细菌或病毒等致病物质[19]。然而,被固定的药物载体也会随着黏液层的更新而被清除,导致转运效率较低。因此,表面为亲水性且电中性的药物载体更容易穿透黏液屏障。
2.1.2 肠上皮细胞屏障小肠黏液下方是由一层连续细胞构成的肠上皮组织,主要包括肠上皮细胞、杯状细胞、潘氏细胞以及M细胞等。相邻的肠上皮细胞间依靠紧密连接相互联系,空隙介于8 ~ 13 Å,维持了肠上皮结构的完整,可防止细菌、毒素等物质进入体内,同时也成为限制药物载体跨膜转运的另一重要物理屏障[20]。由于细胞膜具有亲脂性,且带有负电,因此表面具有亲脂性且带强正电荷的药物载体更易被肠上皮细胞所摄取。然而,这与克服黏液屏障所需具备的载体特性恰好相反,增大了载体设计的难度[21-22]。此外,肠上皮细胞具有一定的极性,因此对于需要进入血液循环并到达靶部位发挥治疗作用的药物而言,载体的胞内转运以及出胞也会影响药物的递送效率。然而,细胞内部含有各种细胞器以及微丝、微管等细胞骨架[23],处于一种高度拥挤的状态,极大地制约了药物载体在胞内的运动[24-25]。
2.1.3 蛋白冠冕作用小肠肠液中存在着大量具有表面活性的分子,如胆盐、消化酶、食物蛋白、磷脂及其降解产物等,这些成分可与药物载体发生相互作用,从而在载体表面形成一层蛋白冠冕[26]。蛋白冠冕的形成会在一定程度上影响药物载体的表面特性(如粒径、电位等),进而影响其功能的发挥。Peng等[27]将阳离子纳米粒(cationic nanoparticles,CNPs)在胃肠液中进行孵育,其中的消化酶吸附在CNPs表面形成了蛋白冠冕,显著降低了其在肠上皮细胞的摄取效率。黏液中的黏蛋白同样是口服药物载体表面蛋白冠冕的主要来源。Yang等[28]证明,金纳米粒表面吸附的黏蛋白会影响其在上皮细胞基底侧的出胞效率。尽管目前针对胃肠道蛋白冠冕的相关研究较少,但是根据已有报道可以发现,蛋白冠冕的形成会显著影响口服药物载体的体内运输过程,这可能是引起其体内外作用效果差异的原因之一。
2.2 蛋白多肽类降糖药物口服载体面临的特殊屏障
胰岛素以及GLP-1类似物必须入血才能发挥降糖作用。因此,药物载体除了需要克服上述共性屏障,还需要解决在肠上皮细胞的胞内转运以及基底侧的出胞屏障,这些屏障直接影响了药物的转运效率,决定其后续功能的发挥。另外,载体经肠上皮细胞转运入血,大部分通过肝门静脉被运送至肝脏。肝脏对于载体以及药物的处置作用也是影响蛋白多肽类降糖药物口服递送的关键因素。
2.2.1 药物载体的胞内转运大多数药物载体经内吞作用进入肠上皮细胞后首先被包裹在囊泡中,然后与内含体发生融合。内含体沿着微管在胞内运动,表面ATP驱动的质子泵不断将H+泵入内腔,使其内部pH由6.5快速降至5.0,并最终与溶酶体发生融合[29-30]。溶酶体内的pH在表面质子泵的作用下可维持在5.0,且内部含有多种酸性水解酶[31]。药物载体经内含体-溶酶体途径被运送至溶酶体中会影响载体结构的完整性从而破坏内部包载的药物,最终影响药物的跨胞转运效率。尤其是针对蛋白多肽类降糖药物口服载体的设计,由于药物在溶酶体内部水解酶的作用下会快速发生降解并失去活性,因此需要对载体进行特殊设计,使其能够发挥溶酶体逃逸作用[32]。此外,还可利用其他胞内转运通路实现药物的高效输运。药物载体经小窝蛋白(caveolin)介导入胞后可不经过溶酶体而直接被运送至内质网或高尔基体[33-34]。高尔基体是胞内蛋白质加工、分拣以及运输的主要部位,可将蛋白质运送至细胞膜或分泌到细胞外。因此,高尔基体/内质网转运途径更利于药物载体在胞内的转运[35]。但是,目前针对口服药物载体胞内转运过程的研究较少,大部分载体在胞内的具体转运机制尚不清楚。
2.2.2 药物载体的出胞载体在肠上皮细胞基底侧的转运效率也是决定蛋白多肽类降糖药物口服生物利用度的关键因素。与入胞过程相反,载体的出胞是一个反向运输的过程,主要由囊泡介导载体从细胞的基底侧排出。胞内的高尔基体/内质网转运通路可以促进载体的高效出胞,载体进入内质网或高尔基体后通过分泌途径经囊泡被运送到细胞外[36]。此外,载体的出胞过程还受到细胞骨架以及动力蛋白的调节,载体在细胞内沿微管进行定向运动有助于其高效出胞[37-38]。尽管目前已提出多种策略来提高载体的入胞效率,但是针对出胞过程的研究却仍较匮乏。同时,大部分口服药物载体存在的“入胞容易,出胞困难”的问题也极大地制约了药物的跨胞转运效率[35]。
综上,药物载体在肠上皮细胞的转运是一个连续的过程,包括入胞、胞内转运以及出胞3个阶段,其中每个阶段都会对载体最终的转运效率产生显著影响。因此,需要逐一克服上述过程才有望实现蛋白多肽类降糖药物在体内的高效输运。
2.2.3 肝脏的处置作用药物需要在靶器官达到有效蓄积,才能精准发挥治疗作用[39]。针对蛋白多肽类降糖药物的口服递送,药物载体高效克服黏液和肠上皮细胞屏障可以确保足够的药物进入血液循环从而发挥降糖作用。但是,一方面药物载体入血后大部分经门静脉被运送至肝脏,肝脏对药物载体的清除作用是影响其体内功能发挥的重要因素。据报道,进入血液中的药物载体约有30% ~ 99%会被肝脏的库普弗细胞(Kupffer cell)摄取,从而被快速清除[40-41]。同时,肝脏也是胰岛素清除的主要场所,由胰岛β细胞分泌的胰岛素主要在肝脏被降解,避免了外周高胰岛素血症所引起的低血糖等不良反应。另一方面,肝脏还是胰岛素作用的靶器官。胰岛素作用于肝脏上的胰岛素受体(insulin receptor,IR)可促进肝脏对葡萄糖的摄取,并将其转化为肝糖原储存起来。据统计,血液中约1/3的葡萄糖被肝脏摄取利用,与肌肉和脂肪组织的摄取量相当[42-43]。因此,肝脏对于蛋白多肽类降糖药物尤其是胰岛素的口服递送至关重要。但是,目前研究人员主要关注于如何设计可高效克服胃肠道屏障的口服药物载体,而忽略了载体及药物入血后在肝脏的处置作用,这可能是限制药物发挥降糖作用的主要原因。
3 蛋白多肽类降糖药物口服递送载体的研究进展
针对蛋白多肽类降糖药物口服递送面临的多重屏障,近年来国内外研究人员提出了多种解决策略,用于提高药物载体在胃肠道的转运效率,进而提高药物的口服生物利用度[19]。此外,为了进一步模拟内源性胰岛素的分泌及分布特征,仿生型胰岛素口服递送载体也逐渐受到人们关注,从而更合理地调控糖尿病餐后高血糖水平并改善血糖的利用[44-45]。本部分对目前蛋白多肽类降糖药物口服递送载体的研究进展进行了总结。
3.1 提高胃肠道转运效率的口服递送载体
3.1.1 具有优势物理属性的口服递送载体药物载体的物理属性如尺寸、形状以及刚性等都会对其在小肠的吸收产生显著影响[46-48]。载体的尺寸可以显著影响其在黏液中的运动速度以及细胞摄取效率[49]。Maisel等[50]发现纳米粒尺寸越小,越容易到达黏液的非流动层而延长滞留时间。Banerjee等[51]研究发现粒子的细胞摄取效率与其尺寸成反比,大小为50 nm的纳米粒在人结肠癌细胞(Caco-2细胞)上的摄取效果最好。然而,载体的尺寸也会影响其内部药物的释放。同等条件下载体的粒径越小,比表面积越大,药物释放速度越快,可能导致药物提前泄露[52]。因此,在载体设计过程中需要进行筛选,从而确定最佳尺寸。
载体的形状以及刚性也可显著影响其在黏液及肠上皮细胞的转运效率。近年来,有实验人员证实棒状纳米粒比球状纳米粒在黏液中的运动速度更快,穿透更深,在胃肠道中驻留时间更长[53],且在肠上皮细胞具有更高的摄取效率,约为球状纳米粒的2倍[51]。此外,由于大部分药物载体具有一定的弹性,在与小肠相互作用过程中会发生形变,从而影响载体的体内命运。笔者课题组在前期研究中发现刚度适中的球形纳米粒在生物凝胶的网状结构中可变形为椭球体,并以“旋转-跳跃”的方式快速运动[54]。因此,调节药物载体的刚性也可在一定程度上影响其在小肠的转运效率。Yu等[55]利用磷脂相变温度的差异,设计了具有不同刚性的脂质体。结果显示刚度适中的脂质体(杨氏模量约为15 MPa)在小肠黏液中穿透速度最快,且细胞摄取效率最高。可显著降低糖尿病大鼠的血糖水平,最低达初始水平的50%,胰岛素口服生物利用度被提高至13.65%。Zheng等[56]构建了具有不同弹性的两性离子水凝胶纳米粒,并研究其在小肠的转运效率。体内外实验结果显示纳米粒的弹性增加,其入胞以及出胞效率相应提高,从而促进药物的跨胞转运。其中弹性最强的纳米粒(hard nanoparticles,HNPs),杨氏模量约为165.2 MPa,在糖尿病大鼠体内降糖效果最好,胰岛素口服生物利用度约为15%。
综上,深入探究药物载体的基本物理属性对其在小肠中的摄取及转运效率的影响,可用于指导蛋白多肽类降糖药物口服递送载体的理性化设计,从而高效克服小肠内的吸收屏障,显著提高药物的口服生物利用度。
3.1.2 靶向配体修饰的口服递送载体肠上皮细胞是限制口服药物载体吸收的主要屏障,其中顶侧膜屏障是制约载体入胞的关键因素。因此,为了提高药物的入胞效率,配体介导的靶向递送策略被广泛应用。在载体表面修饰与肠上皮细胞表面的受体或转运体特异性结合的功能化配体,可显著提高载体的细胞摄取效率[57]。然而有研究发现,一些配体修饰的口服递送载体尽管可以高效入胞,但出胞效率极低,即存在着“入胞容易,出胞困难”的现象[58]。针对这一问题,越来越多的研究人员开始关注药物在胞内的转运及出胞过程,并设计了可实现高效跨胞转运的蛋白多肽类降糖药物口服递送载体(见表1[59-65])。

表1 靶向配体修饰的蛋白多肽类降糖药物口服递送载体Table 1 Examples of targeting ligand-modified oral drug carriers for hypoglycemic proteins and peptides
目前该类载体的设计多基于具有明确胞内转运通路的物质,其中免疫球蛋白G(immunoglobulin G,IgG)是体内最主要的抗体,由肠道中高表达的新生儿Fc受体(neonatal Fc receptor,FcRn)介导转运[66]。FcRn以pH依赖的方式与IgG的Fc部分进行结合,主要表现为在pH低于6.5时具有较高亲和力,而在中性环境(pH 7.4)下结合力较低[67-68]。因此,利用FcRn与IgG的结合特性,通过在载体表面修饰IgG的Fc片段可以促进其跨胞转运。Pridgen等[64]设计的表面修饰Fc片段的纳米粒(nanoparticles-Fc,NPFc),在酸性的胃肠道环境下通过与肠上皮细胞表面的FcRn特异性结合而被摄取入胞。NP-Fc与FcRn形成的复合物在胞内主要通过内含体/溶酶体途径进行转运,酸性条件下二者之间的高亲和力在一定程度上降低了溶酶体的降解作用[69]。出胞后在中性环境下NP-Fc与FcRn发生解离并最终进入血液循环发挥作用。结果表明Fc可高效介导纳米粒的跨胞转运,NP-Fc在体内的吸收效率约为未修饰纳米粒的10倍。Shi等[70]制备的Fc修饰的艾塞那肽纳米粒也显示了较强的跨胞转运能力,且经口服给药后可持续发挥降血糖作用(长达12 h)。人体内的胆酸主要由位于回肠细胞顶端的钠离子依赖型胆酸转运体(apical sodium-dependent bile acid transporter,ASBT)介 导转运[71]。近年来有研究表明,载体表面修饰胆酸或脱氧胆酸可显著提高胰岛素的口服生物利用度。Fan等[65]制备了表面修饰脱氧胆酸的壳聚糖纳米粒(deoxycholic acid-modified nanoparticles,DNPs),DNPs与Caco-2细胞表面的ASBT结合后被摄取入胞,并进一步破坏溶酶体膜结构发挥逃逸作用。胞质中的DNPs可与回肠胆酸结合蛋白(ileal bile acid-binding protein,IBABP)相结合从而被运送至细胞基底侧。通过利用肠道胆酸转运通路,DNPs将胰岛素的跨胞转运效率提高了12.86倍,最终胰岛素的口服生物利用度可达15.9%。Gao等[72]制备的表面修饰脱氧胆酸的介孔硅纳米粒,同样可促进胰岛素在小肠的渗透,在糖尿病大鼠上发挥显著的降血糖作用。通过修饰特异性靶向配体来提高载体在肠上皮细胞的转运效率是最常见也是最简单的策略。但是,目前大部分的研究主要关注载体的入胞效率,而对载体的胞内命运及出胞机制研究较少。因此需要深入探究药物载体在胞内的转运过程,阐明配体修饰与载体胞内转运通路的相关性,从而用于指导具有高效跨胞作用的药物载体的设计,最终达到提高蛋白多肽类降糖药物口服生物利用度的目的[35]。
3.1.3 表面性质可转换的口服递送载体针对黏液屏障与肠上皮细胞屏障间存在的递药矛盾,可通过调节药物载体表面不同功能配体的长度[73]或载体的亲/疏水性[74],从而筛选出跨肠道屏障转运效率最高的递送策略。然而,这种方法需要同时制备多种载体并对其性质进行考察,因此耗时长、成本高且较复杂。与之相比,设计表面性质可变的口服递送载体则更为简单可行,即在黏液中载体表面为高亲水性以及电中性,减少与黏蛋白间的相互作用,快速穿透黏液屏障;到达细胞表面后则暴露出载体内部的正电性,有利于其被肠上皮细胞所摄取。
Wang等[75]利用蛋白冠冕作用,预先在阳离子脂质体(cationic liposomes,CLs)表面吸附了一层牛血清白蛋白(bovine serum albumin,BSA)制备了内部包载胰岛素的蛋白冠冕阳离子脂质体(protein corona cationic liposomes,PcCLs)。吸 附BSA后PcCLs整体带少量负电荷(约为-10 mV),有助于其在黏液中快速运动,同时BSA在小肠内蛋白酶的作用下会逐渐发生降解并暴露出内部带强正电的CLs,从而促进胰岛素在肠上皮细胞的摄取。此外,还可利用肠道微环境响应性地触发载体表面性质的转变,如Wu等[76]在聚乳酸-羟基乙酸共聚物[poly(lactic-co-glycolic acid),P]纳米粒表面同时修饰了阳离子八聚精氨酸(octa-arginine,R8)和阴离子磷酸丝氨酸(phosphoserine,Pho),制备了具有电荷翻转能力的仿生纳米粒P-R8-Pho。P-R8-Pho可模拟病毒在黏液中快速运动,接着在小肠碱性磷酸酶(intestinal alkaline phosphatase,IAP)作用下特异性水解表面的Pho,暴露出内部的R8从而介导纳米粒高效穿过肠上皮细胞屏障。P-R8-Pho的酶响应性电荷翻转作用可以显著提高胰岛素在小肠的转运能力,口服生物利用度达5.96%。通过多种手段调节表面性质的转变,载体可依次高效克服黏液屏障以及肠上皮细胞屏障,有效提高蛋白多肽类降糖药物的口服生物利用度,并为后续研究打下基础。
3.1.4 其他新型口服递送载体除了上述策略以外,科学家们还设计了许多新型蛋白多肽类降糖药物的口服递送载体,它们在活体动物水平都显示了良好的降血糖作用,并显著提高了药物的口服生物利用度。离子液体是由有机阳离子和无机或有机阴离子组成的一类液体盐,熔点通常在100 ℃以下,被广泛用于化学研究和制剂技术[77]。Banerjee等[78]利用胆碱和香叶酸离子液体(choline and geranate,CAGE)制备了高效口服胰岛素制剂。CAGE可保护胰岛素免受胃肠道内酶的降解,并降低小肠黏液的黏度,促进胰岛素在黏液中的渗透。CAGE主要通过打开细胞间紧密连接,显著提高胰岛素在肠上皮细胞的转运效率,约为对照组的10倍。进一步将CAGE与胰岛素形成的复合物包裹进肠溶胶囊中,其在糖尿病大鼠上显示了持续的降血糖作用(约达12 h),胰岛素的口服生物利用度可提高至51%。
病毒衣壳表面分布着同等密度的正负电荷(净中性),且具有较强亲水性,有助于病毒快速穿透黏液屏障实现高效侵袭[79]。受病毒这一表面特性的启发,近年来部分研究人员利用两性离子制备药物载体用于高效克服肠道吸收屏障。Han等[80]将甜菜碱聚合物(polycarboxybetaine,PCB)与1,2-二硬脂酰基-sn-丙三基-3-磷脂酰乙醇胺(1,2-distearoylsn-glycero-3-phosphoethanolamine,DSPE)相连,制备了一种两性离子甜菜碱聚合物胶束DSPE-PCB。DSPE-PCB可模拟病毒衣壳表面高电荷密度且电中性的特性,快速穿过小肠黏液屏障,在黏液中运动的均方位移(mean square displacement,MSD)约为黏液穿透型纳米粒(含有聚乙二醇链的聚山梨酯胶束)的12倍。PCB是细胞表面质子辅助氨基酸转运体1(proton-assisted amino acid transporter 1,PAT1)的底物,可介导DSPE-PCB穿过肠上皮细胞屏障,在不打开细胞间紧密连接的前提下将胰岛素高效递送入血。将冻干后DSPE-PCB包裹进肠溶胶囊中,可有效降低糖尿病大鼠的血糖水平,胰岛素口服生物利用度达42.6%。
针对GLP-1类似物的口服递送,由于其结构以及作用方式与胰岛素存在差异,因此在载体设计上也需要进行相应的改进。Lin等[81]制备了内部包载艾塞那肽(exenatide,EXT)的相转变纳米乳(phasechangeable nanoemulsions,EXT@PC/NEMs)。随着温度升高,构成纳米乳的脂肪酸由固态变为液态。因此,在人体温度下EXT@PC/NEMs的可变形性增强,细胞摄取及转运效率相应提高,可促进艾塞那肽在小肠的吸收。EXT@PC/NEMs主要经小肠淋巴系统转运到达胰腺,并释放艾塞那肽作用于胰岛α和β细胞,从而促进胰岛素的分泌,抑制糖原降解,有效控制2型糖尿病大鼠的血糖水平,最终艾塞那肽的相对生物利用度达23.8%。Xu等[82]设计了一种含有反胶束的脂质纳米胶囊(lipid-based nanocapsules containing reverse micelles,RM-LNC),该纳米胶囊既可用于口服递送艾塞那肽,也可促进L细胞分泌GLP-1,2种功能协同作用在2型糖尿病小鼠上显示了良好的降血糖作用。同时,长期给药还可改善小鼠体内葡萄糖代谢,减轻胰岛素抵抗。由于GLP-1及其类似物在体内易被降解,且主要在胰腺发挥作用,因此需要提高药物载体在体内的循环时间,增加胰腺靶向能力,从而使药物更好地发挥降糖作用。
近年来,可实现体内注射的新型口服给药装置也被广泛应用到蛋白多肽类降糖药物的递送中。麻省理工学院的研究学者研发了一种腔内展开微针注射器(luminal unfolding microneedle injector,LUMI)口服胶囊。当LUMI到达小肠后外部的胶囊发生降解,内部的3条折叠臂会弹开,其上的微针会将装载的胰岛素直接注射入小肠壁中。作用结束后折叠臂发生断裂并被排出体外,不会引起肠阻塞等不良反应。动物实验结果表明:LUMI比皮下注射胰岛素起效更快,在4 h内全身吸收效率达10%[83]。另外,科学家们还通过仿生豹纹龟的龟壳设计了一种自主定向毫米级涂药器(self-orienting millimeter-scale applicator,SOMA)。SOMA内部是一个由弹簧连接的载有胰岛素的微针。该装置被吞入体内后,可在胃部快速调整为直立状态以保证底部紧贴胃壁,随后微针直接插入胃壁组织,将胰岛素释放入血。体内结果显示,口服SOMA可产生与皮下注射胰岛素相似的降血糖作用,且血中胰岛素水平相当[84]。上述新型口服给药装置的面世从物理学的角度为蛋白多肽类降糖药物口服递送载体的设计提供了新的思路。
3.2 仿生型胰岛素口服递送载体
3.2.1 肝脏靶向型口服胰岛素递送载体肝脏是内源性胰岛素的主要作用部位,正常生理状态下体内胰岛素在肝脏与外周间存在浓度梯度。因此,设计肝脏靶向的口服胰岛素载体可以模拟内源性胰岛素的分布,提高肝脏处胰岛素的蓄积,从而有效发挥降血糖作用。Zhang等[85]设计了一种肝脏靶向的口服胰岛素纳米粒,通过将胆酸修饰在纳米粒表面,其可经肝肠循环途径靶向至肝脏,从而有效发挥降血糖作用,胰岛素的相对药效学生物利用度被提高至26.9%。此外,美国Diasome公司研发的口服肝靶向胰岛素囊泡(hepatic-directed vesicle insulin,HDV-Ⅰ)可将胰岛素选择性输送至肝脏,有效控制糖尿病患者的餐后血糖水平。但是,目前针对具有肝脏特异性靶向功能的口服胰岛素载体的报道仍然较少,而更多关注于注射用肝靶向胰岛素的研究。Shao等[9]设计的胰岛素原-转铁蛋白复合物,皮下注射给药可以模拟内源性胰岛素在肝脏的选择性作用,抑制肝糖原的降解并持续发挥降血糖作用(长达40 h),同时避免了由于外周胰岛素浓度过高而产生的低血糖等不良反应。由此可见,胰岛素选择性作用在肝脏可发挥良好的降血糖作用,肝脏靶向型胰岛素为胰岛素的口服递送提出了新的研究方向,有望给糖尿病的治疗带来新的启发。
3.2.2 葡萄糖响应型口服胰岛素递送载体在正常生理状态下,血液中葡萄糖水平升高会促进胰岛β细胞分泌,从而将含有胰岛素的囊泡释放入血发挥降糖作用,因此胰岛素的释放主要受体内血糖水平的调控[86]。但是,目前大部分的胰岛素口服递送载体主要通过在体内的降解释放药物,无法精准调控胰岛素的释放速度以及释放量,可能会引起低血糖。同时,由于个体间吸收差异较大,还可能出现血糖控制不平稳的现象,导致血糖波动过大,这也是引起糖尿病并发症的主要原因[87]。因此,如何根据体内血糖变化精准调控胰岛素的释放成为口服胰岛素载体设计的一大挑战。
Yu等[45]制备了一种葡萄糖响应型口服胰岛素载体。载体表面是由苯硼酸-透明质酸聚合物(hyaluronic acid-phenylboronic acid,HA-PBA)形成的外壳,内部为表面修饰Fc的胰岛素脂质体。当血糖升高时,苯硼酸与葡萄糖结合导致外壳分离,暴露出内部的脂质体核心。脂质体表面的Fc配体可进一步促进其在肠上皮细胞的摄取及转运。实验结果显示,当葡萄糖浓度达10 mmol · L-1时即可触发脂质体表面HA-PBA的脱落,从而响应性降低糖尿病大鼠餐后高血糖水平,且不会引起低血糖反应。Wang等[44]利用过氧化氢敏感的两亲性聚合物——聚乙二醇-聚甲硫氨酸(polyethylene glycolpolymethionine,PEG-PolyMet)制备了一种神经节苷脂-单唾液酸靶向肽(ganglioside-monosialic acid-targeting peptide,简称Pep)修饰的聚合物囊泡(Pep-polymersomes,Pep-PMS),其内部包载葡萄糖氧化酶(glucose oxidase,GOx)以及胰岛素。Pep-PMS具有葡萄糖响应智能释药的特性。在高糖环境下,GOx氧化葡萄糖产生过氧化氢,可将PEG-PolyMet中的疏水性侧链转变为亲水性,从而使囊泡破裂并释放内部胰岛素。体内外结果显示Pep-PMS可通过被动靶向作用蓄积在肝脏的窦周隙,并响应体内高血糖选择性释放胰岛素。Pep-PMS在糖尿病大鼠体内显示了持续的、响应性的降血糖作用,并显著提高了肝糖原的合成。
综上,基于内源性胰岛素的分泌以及分布模式,仿生设计的口服胰岛素递送载体可以精准调控餐后高血糖水平,恢复肝脏-外周的胰岛素浓度梯度,改善血糖的利用,为口服胰岛素递送载体的设计带来新的理念。
4 蛋白多肽类降糖药物口服递送载体的临床研究进展
随着对蛋白多肽类降糖药物口服递送载体研究的不断深入,现已有产品上市或进入临床研究阶段,根据其递送策略的不同可简单划分为3类:1)吸收促进剂类,如丹麦Novo Nordisk制药公司开发的口服索马鲁肽片剂以及以色列Oramed制药公司和合肥天麦生物联合研发的口服胰岛素胶囊ORMD-0801[88-89]等;2)微纳米载体类,如美国Diasome公司研发的口服肝脏靶向的胰岛素囊泡HDV-I以及以色列Oshadi制药公司研发的口服胰岛素纳米粒Oshadi-Icp[90-91]等;3)新型给药系统类,如美国Rani制药公司设计并开发的“机器人”药丸RaniPillTM等。这些制剂在临床研究中显示了良好的降血糖作用,口服生物利用度约达1%左右,具有一定的临床应用前景。其中,由Novo Nordisk开发的口服索马鲁肽片剂(商品名:Rybelsus®)于2019年9月经美国食品药品监督管理局(Food and Drug Administration,FDA)批准上市,这是全球首个获批上市的口服GLP-1类似物,极大地推动了蛋白多肽类降糖药物口服制剂的研发进程,具有里程碑式的重要意义[92]。
吸收促进剂可以提高肠上皮细胞的通透性从而促进药物的口服吸收,主要作用机制包括直接打开细胞间紧密连接或提高细胞膜的流动性[93]。由于制备工艺简单,吸收促进剂被广泛应用于蛋白多肽类降糖药物口服制剂的开发中,临床效果良好,具有一定的应用前景。索马鲁肽是一种长效GLP-1类似物,相对分子质量为4 113,体内半衰期长达165 h[94]。Novo Nordisk利用Eligen®技术,即添加一种小分子吸收促进剂N-[8-(2-羟基苯甲酰基)氨基]辛酸钠[sodiumN-[8-(2-hydroxybenzoyl)amino]caprylate,SNAC]制备了索马鲁肽片剂。与大多数口服药物制剂在肠道吸收不同,索马鲁肽片剂主要在胃部吸收[6](见图2[95]),且吸收程度与SNAC的用量相关,体内研究表明300 mg的SNAC可产生最高的索马鲁肽血浆浓度。SNAC可以帮助维持索马鲁肽的单体形式,并且通过缓冲作用升高胃内局部pH,提高索马鲁肽的溶解度,防止其被胃内的蛋白酶降解。此外,SNAC具有较强的亲脂性,通过与胃上皮细胞相互作用,可促进索马鲁肽在胃部的快速吸收[6,96]。在SNAC多种促吸收机制的作用下,索马鲁肽在体内的生物利用度可达1.22%[97]。临床试验结果显示,口服索马鲁肽具有较显著的降糖减肥作用,可有效控制2型糖尿病患者的血糖水平,且作用效果显著优于恩格列净和西格列汀等小分子降血糖药物[98-99]。口服索马鲁肽的上市有望改善糖尿病患者的用药方式,提高病人的生活质量,从根本上解决皮下注射给药方式带来的不便。

图2 口服索马鲁肽吸收机制[95]Figure 2 The absorption mechanism of oral semaglutide[95]
Oramed制药公司开发的新型蛋白多肽类药物口服递送技术PODTM外部为肠溶胶囊,内部含有乙二胺四乙酸(ethylenediaminetetraacetic acid,EDTA)和胆盐等吸收促进剂,可以通过打开细胞间紧密连接促进药物在小肠的吸收。此外,胶囊内部还含有大豆胰蛋白酶抑制剂和抑肽酶等,保护药物不被胃肠道内的蛋白酶降解,维持药物稳定性[100]。合肥天麦生物与Oramed公司利用PODTM技术联合开发的口服胰岛素肠溶胶囊ORMD-0801,Ⅱ期临床试验结果显示可有效降低1型糖尿病患者的餐后血糖以及2型糖尿病患者的空腹血糖且安全性良好[101]。2020年,ORMD-0801获得中国国家药品监督管理局(National Medical Products Administration,NMPA)的批准,进入中国开展Ⅲ期临床试验,给胰岛素口服制剂的研究带来新的曙光。
尽管在基础研究中多利用微纳米载体克服胃肠道屏障,促进蛋白多肽类降糖药物的口服吸收,以达到较高的生物利用度,但受限于复杂的制备工艺以及较高的生产成本,其中仅有少量进入临床研究阶段。由美国Diasome公司研发的HDV-I,其磷脂双分子层中含有肝细胞靶向分子(hepatocytetargeting molecule,HTM)和生物素磷脂酰乙醇胺(biotin-phosphatidylethanolamine,biotin-PE),可模拟生理条件下的门静脉胰岛素灌注,将胰岛素选择性输送至肝脏从而重建肝脏对葡萄糖的代谢调节作用,促进肝脏对葡萄糖的摄取、提高肝糖原的储存并有效降低血糖[102]。临床试验结果显示口服HDV-I可有效控制1型和2型糖尿病患者的餐后血糖水平,且不引起低血糖等不良反应的发生[103-104],目前HDV-I正处于Ⅲ期临床研究阶段。
此外,美国Rani制药公司设计并开发了一种新型给药系统——RaniPillTM,这是一种微型注射胶囊,表面为肠溶包衣,可以保护胶囊在胃部不被破坏。在小肠中,由于pH升高,肠溶衣溶解,胶囊内部的气球充气膨胀并通过压力将含有药物的微针直接推入肠壁,残余的装置则在几天内排出体外。Ⅰ期临床研究显示,RaniPillTM胶囊在体内安全性良好,不会引起不良反应,且不受食物的影响。该胶囊有望突破生物药口服瓶颈,成为胰岛素口服给药的新载体[105]。表2总结了绝大部分口服蛋白多肽类降糖药物制剂的研发概况。
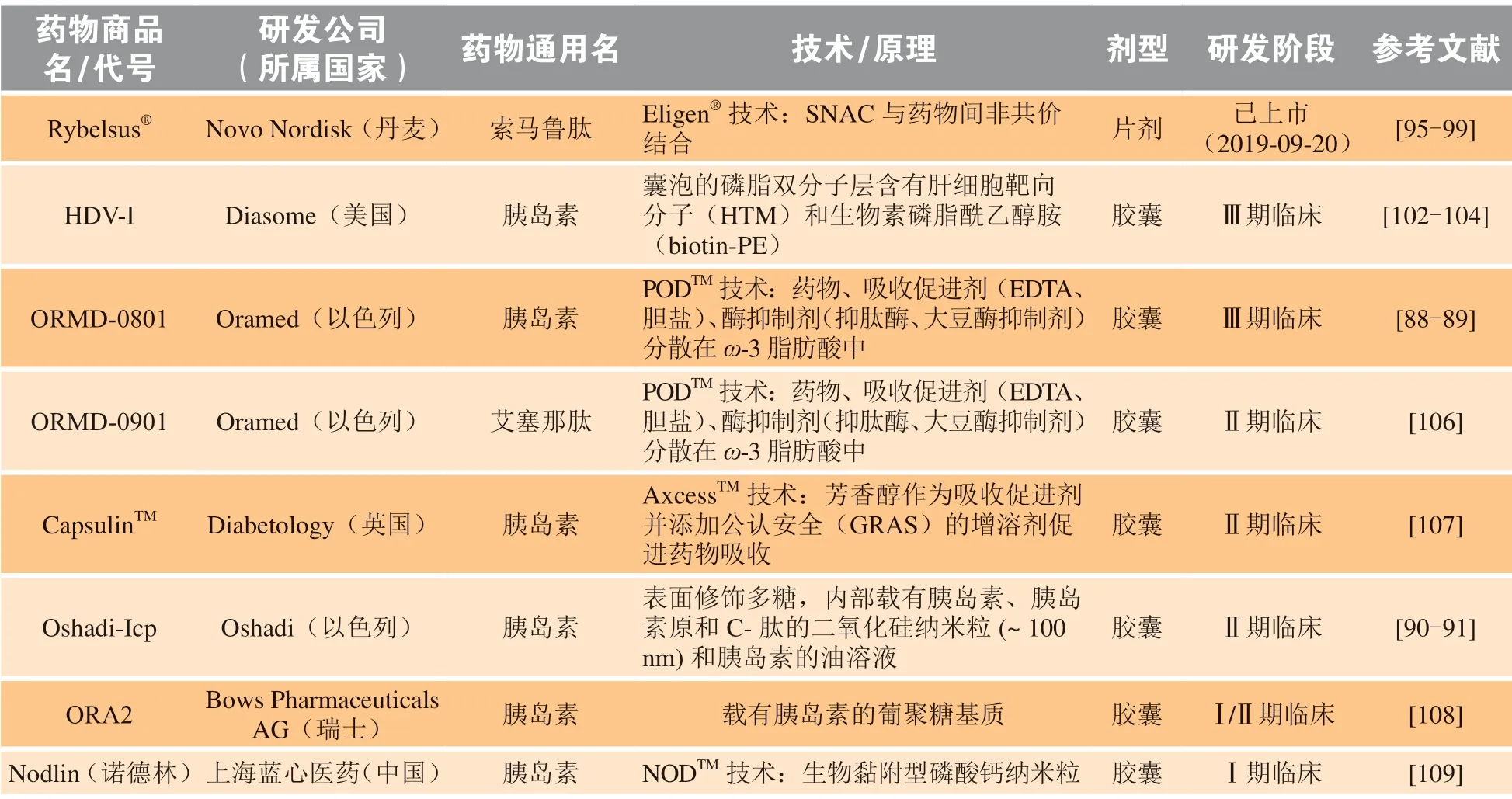
表2 已上市或处于临床研究阶段的蛋白多肽类降糖药物口服制剂Table 2 Oral preparations of hypoglycemic proteins and peptides on the market or in clinical research
5 总结及展望
蛋白多肽类降糖药物口服递送载体的设计是近年来的研究热点。尽管目前相关研究已有较多报道,然而这一领域仍面临着许多挑战。一方面,基础研究中多关注于如何克服胃肠道的生物化学屏障(如pH、酶等)以及肠上皮细胞的顶侧膜屏障,并未有效解决载体“入胞容易,出胞困难”这一关键科学问题。此外,由于糖尿病患者间存在个体差异,如何更精准地调控药物释放,减轻患者的血糖波动成为蛋白多肽类降糖药物口服递送载体设计的更高需求。另一方面,目前临床上蛋白多肽类降糖药物口服制剂研发进程缓慢,且多集中于开发安全高效的口服吸收促进剂以提高药物的吸收效率。尽管口服索马鲁肽已成功上市,但其生物利用度仍较低,仅为1% ~ 2%。因此,开发新型制剂技术显著提高药物的口服生物利用度,使其突破给药方式的限制,更高效地发挥降糖作用成为蛋白多肽类降糖药物口服制剂临床转化的主要目标。最后,该领域还存在着基础研究与临床应用严重脱节,产出比低下的问题。主要是目前基础研究中大多利用微纳米载体促进蛋白多肽类降糖药物的口服吸收,在小动物模型上通常可以达到较高的口服生物利用度,但是缺少在大动物模型如比格犬体内的药效验证,难以评估其在人体内的疗效。同时,受限于复杂的制备工艺以及较高的生产成本,大部分药物载体的临床转化效率较低。因此,开发经济高效、制备简单且质量可控的口服药物载体有助于推动其临床转化进程。
综上,尽管蛋白多肽类降糖药物的口服递送面临着重重困难,但是其对于糖尿病的临床治疗显示了重要的意义,有望提高糖尿病患者的用药依从性,改善生存质量。虽然目前大部分的研究仍处于临床前研究阶段,临床转化效率较低,但是相信随着载体材料和制剂技术的不断发展、各种新型口服吸收促进剂的相继提出以及精准医学时代的到来,开发出一系列安全、高效的蛋白多肽类降糖药物口服制剂将成为可能。
