氧弹燃烧量热耦合离子色谱法分析危废热值和非金属元素
2022-06-27蒋祺东方薇瑶杨微微张莉陈双伟潘时俊
*蒋祺东 方薇瑶 杨微微 张莉 陈双伟 潘时俊
(1.绍兴凤登环保有限公司 浙江 312074 2.浙江凤登绿能环保股份有限公司 浙江 321100)
1.概述
危险废物主要采用焚烧或裂解气化技术处置,后者更有利于危险废物资源化。例如水煤浆气化炉协同处置固体废物,在处置危险废物同时,将其中碳、氢、氮、硫和氯等元素转化为碳铵、氢气、液氨、硫磺和工业盐等产品,玻璃体的炉渣可作为建筑材料。在危废裂解工艺中,危废热值及其氟、氯、溴和硫含量是入炉物料配伍的关键指标。如控制不当,严重影响气化炉稳定运行,轻则造成停工停产,重则造成生产安全事故。
目前,危废处置企业主要采用氧弹燃烧量热仪分析危废热值,燃烧物水稀释后定容,再分别采用氟选择离子电极、定硫仪和氧化还原滴定法定量分析氟、硫和氯(溴)。其中滴定法不能区分氯和溴,两者共存时相互干扰。分项方法操作繁琐、干扰因素多、人为影响大,直接影响分析结果的随机误差。原理上,多种仪器分析法可一次分析试样中氟、氯、溴、硫及其它元素,例如波长色散X射线荧光光谱(XRF)[1]、X射线吸收近边缘结构光谱(XANES)[2]、电感耦合等离子光谱仪(ICP)[3]、电感耦合等离子质谱仪(ICPMS)[4]和燃烧裂解离子色谱法(C-IC)[5~7]。然而,应用XRF或XANES方法,因危废成分组成复杂和不确定性,不易制备理想的通用标准样条,一般仅适合定性或半定量分析。ICPS或ICPMS方法一般采用消解分解处理试样,通用消解方法常使用盐酸、硫酸、硝酸甚至氢氟酸,从而干扰样品中氟、氯和硫分析。IC法方面,采用氧气流中燃烧裂解危废试样后离子色谱法分析氯、溴和硫[7]。当有机危废蒸气压相对较大时,该方法易在危废燃烧分解前部分气化被载气带出系统而导致元素含量分析结果偏低。近代发展的C-IC技术将燃烧裂解炉与离子色谱仪联用,实现燃烧裂解、试样转移和定容和离子色谱分析的自动控制,一些蒸气压较高的有机物在燃烧分解前可能部分气化,以分子态进入吸收液或被载气带出系统,导致离子色谱分析回收率降低。另一方面,分解温度较高的无机物因分解不完全而残留于裂解炉,同样降低回收率。此外,裂解过程易积焦或积炭,分析元素的完全转移存在不确定性。
上述各类方法除ICP(MS)外的工作原理均不适合与氧弹燃烧量热法耦合分析危废热值,而氧弹燃烧量热耦合离子色谱法则较为可行,但需考虑消除/减小因危废燃烧产物引起的干扰,以及解决危废被测元素含量范围往往超出色谱分析线性范围的问题,从而提高分析方法的通用性。有专利[8]和文献[9~11]报道氧弹燃烧-离子色谱分析卤素和硫方法,基本步骤是水稀释氧弹燃烧产物并定容后直接离子色谱分析。在危废燃烧产物水溶液存在分散相时,因分散相微粒可吸附待测离子化合物,导致离子色谱分析误差增大,仅适应氧弹燃烧产物能形成较为均质水溶液的危废。有研究[12]认为氧弹燃烧危废过程产生较多碳酸盐,而碳酸根对离子色谱分析卤素离子干扰较大,提出采用强酸性离子交换树脂处理,以提高分析结果可靠性。但该法能否抑制因分散微粒的吸附作用引起干扰还不确定。同时,离子交换树脂处理样品是相对复杂的操作过程,易增加随机误差。本文探索更便捷方法抑制危废燃烧液分散相引起的干扰,并结合二项式标线应用实现通用性更好的氧弹燃烧量热-离子色谱分析方法,并通过分析方法学研究验证其可靠性。
2.实验方法
(1)仪器试剂
瑞士万通ECO IC型离子色谱仪,Metrosep A Supp5-150/4.0色谱柱;湖南友欣YX-ZR9703型氧弹燃烧量热仪,气体吸收装置;湖南湘仪实验室仪器开发有限公司H1850型离心机;坛墨质检产1000mg/L离子单标;自制蒸馏水;化学试剂除特殊说明外均为分析纯。
(2)热值分析
危废热值分析操作步骤为:步骤1:带支架和电极的不锈钢氧弹内放入坩埚并加满蒸馏水,超声洗涤4~5min,倒出清洗液。沿氧弹内壁加入10mL浓度为0.1mol/L的NaOH水溶液,充分湿润氧弹内壁。步骤2:依次精确称取0.2g试样和1.0g甲醇于不锈钢坩埚后置于氧弹支架,安装点火丝后将支架放入氧弹筒内,拧紧氧弹筒盖。步骤3:调节氧气钢瓶减压阀控制表压缓慢增加至3.0MPa,维持30s完成氧弹充氧。步骤4:氧弹置于量热仪升降架上,输入相关参数后启动量热仪,量热仪自动运行并记录数据,约20min完成分析。
(3)样品收集与转移
完成危废热值分析后,按以下操作步骤制备分析试样。
步骤1:氧弹置于气体吸收装置上,调节放气阀在~30min释放完氧弹内未凝气体,200mL蒸馏水吸收其中水溶性气体。步骤2:打开氧弹盖,用步骤1的吸收液分三次淋洗氧弹内壁、坩埚、支架和氧弹盖,淋洗液合并于氧弹内,并将坩埚和支架放入氧弹内,超声3min后转移至500mL广口瓶中。另用200mL蒸馏水同法洗涤,洗涤液合并于广口瓶中。步骤3:盛有洗涤液的广口瓶置于70~80℃水浴,间隙摇动或玻棒搅动。2~4h后取出广口瓶,静置待温度降至室温后离心沉降,清液转移至500mL容量瓶。另用80mL蒸馏水分两次洗涤广口瓶,两次的洗涤液均用于离心沉降法洗涤沉降物,洗涤液合并于容量瓶,蒸馏水定容。
(4)离子色谱分析
①离子色谱操作条件。流动相为1mmol/L NaHCO3+3.2mmol/L Na2CO3,流量1.4mL/min。再生液为100mmol/L H2SO4,流量0.7mL/min。10μL定量环,一次性进样器进样20mL。该色谱条件下各离子分离良好,图1和图2分别是氟、氯、溴和硫混标和实际危废离子色谱图,其中未标示保留时间色谱峰是硝酸根。

图1 混标离子色谱图

图2 实际危废色谱图
②混标线制作。移液管移取一定体积的1000mg/L氟、氯、溴和硫酸根离子单标溶液于同一容量瓶中,蒸馏水定容后配制成[F−]、[Cl−]、[Br−]和[SO42−]分别为50.00mg/L、100.00mg/L、50.00mg/L和100.00mg/L的起始混标液。蒸馏水逐级稀释该起始混标液配制成[F−]=0.05mg/L、0.25mg/L、0.50mg/L、1.25mg/L、2.50mg/L、5.00mg/L、12.50mg/L、25.00mg/L、37.50mg/L,[Cl−]=0.10mg/L、0.50mg/L、1.00mg/L、2.50mg/L、5.00mg/L、10.00mg/L、25.00mg/L、50.00mg/L、75.00mg/L,[Br−]=0.05mg/L、0.25mg/L、0.50mg/L、1.25mg/L、2.50mg/L、5.00mg/L、12.50mg/L、25.00mg/L、37.50mg/L及[SO42−]=0.10mg/L、0.50mg/L、1.00mg/L、2.50mg/L、5.00mg/L、10.00mg/L、25.00mg/L、50.00mg/L、75.00mg/L的系列混标溶液。按浓度低至高顺序离子色谱分析混标液,[F−]、[Cl−]、[Br−]和[SO42−]与对应色谱峰面积的线性回归或二次项拟合线作为标准曲线。
③色谱分析。离子色谱分别分析空白和上述第三步的定容液,选定标准曲线计算定容液中氟、氯、溴和硫酸根离子浓度。并按下式计算危废样品的氟、氯、溴、硫元素百分含量:
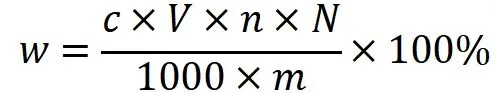
其中,w为危废相关元素重量百分含量,%;c为定容液中离子浓度,mg/L;V为定容体积,L;m为危废样品称量,g;n为定容液中离子浓度超出标线最大浓度后的稀释倍数;N为离子对应元素质量的转换系数。
3.结果与讨论
(1)热值分析。按2.2节方法分析不同模拟危废和实际危废热值,考察所建方法的热值分析误差和偏差,结果分别列于表1和表2。

表1 苯甲酸、DMF水溶液和DMSO水溶液热值相对误差(RE)与相对标准差(RSD)(n=5)

表2 实际危废热值分析相对标准差(RSD)(n=5)
表1所列模拟危废热值分析结果可见,方法分析含水量不高有机化合物热值的相对误差小于3%,相对标准差小于4%,分析结果正确度和精度均能满足要求。水含量增加,分析结果相对误差和均表现出增大趋势,但水干扰氧弹燃烧量热仪热值分析是普遍存在的。表2所列结果可见,分析实际危废热值相对标准差小于3%,表明方法应用于不同热值实际危废分析重复性良好。
(2)检测器线性范围。按2.4.2节方法配制系列混标溶液,2.4.3节方法离子色谱分析混标液,所得各离子不同浓度对应峰面积线性拟合曲线列于图3。
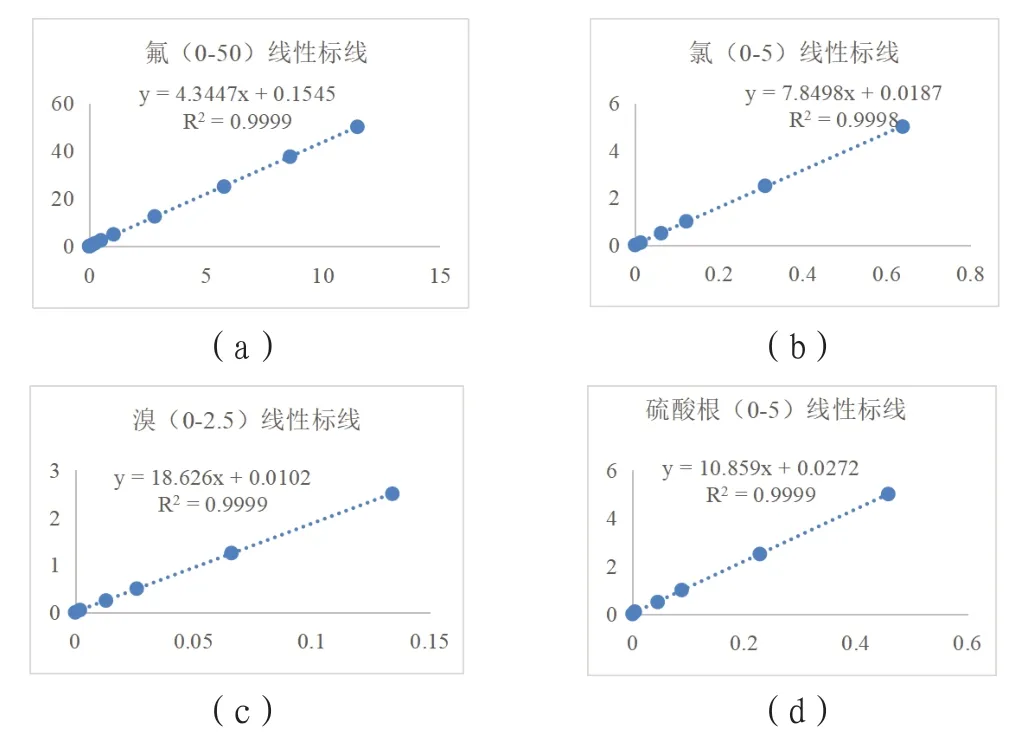
图3 各离子不同浓度对应峰面积线性拟合曲线图
各离子的C~A采用最小二乘法线性回归,其中氟在实验浓度范围内线性相关系数R2=0.9999,检测器线性范围0~50mg/L。而[Cl−]、[Br−]和[SO42−]则分别在0.00~5.00mg/L、0.00~2.50mg/L和0.00~5.00mg/L的线性回归线相关系数R2均接近1(R2>0.9996),截距b均接近0(b<0.04)。而进一步扩大离子浓度范围,R2值偏离1或b值偏离0明显增加。由此,氯、溴和硫酸根离子线性范围分别为0.00~5.00mg/L、0.00~2.50mg/L和0.00~5.00mg/L。
(3)离子色谱分析标线。危废元素含量范围宽,按3.1和3.2节操作的试样溶液离子浓度可能超出检测器线性范围。实验发现氯、溴和硫酸根离子浓度分别在5.0~100.0mg/L、2.50~50.0mg/L和5.00~100.0mg/L,即在检测器线性范围外时,C~A回归线的线性相关系数偏离1程度增加,作为标线的计算误差随之增大。为减小计算误差并提升方法通用性,试用二项式拟合曲线为标线解决该问题,图4为氯、溴和硫酸根离子在5.0~100.0mg/L、2.50~50.0mg/L和5.00~100.0mg/L浓度范围的线性拟合曲线和二项式拟合曲线。
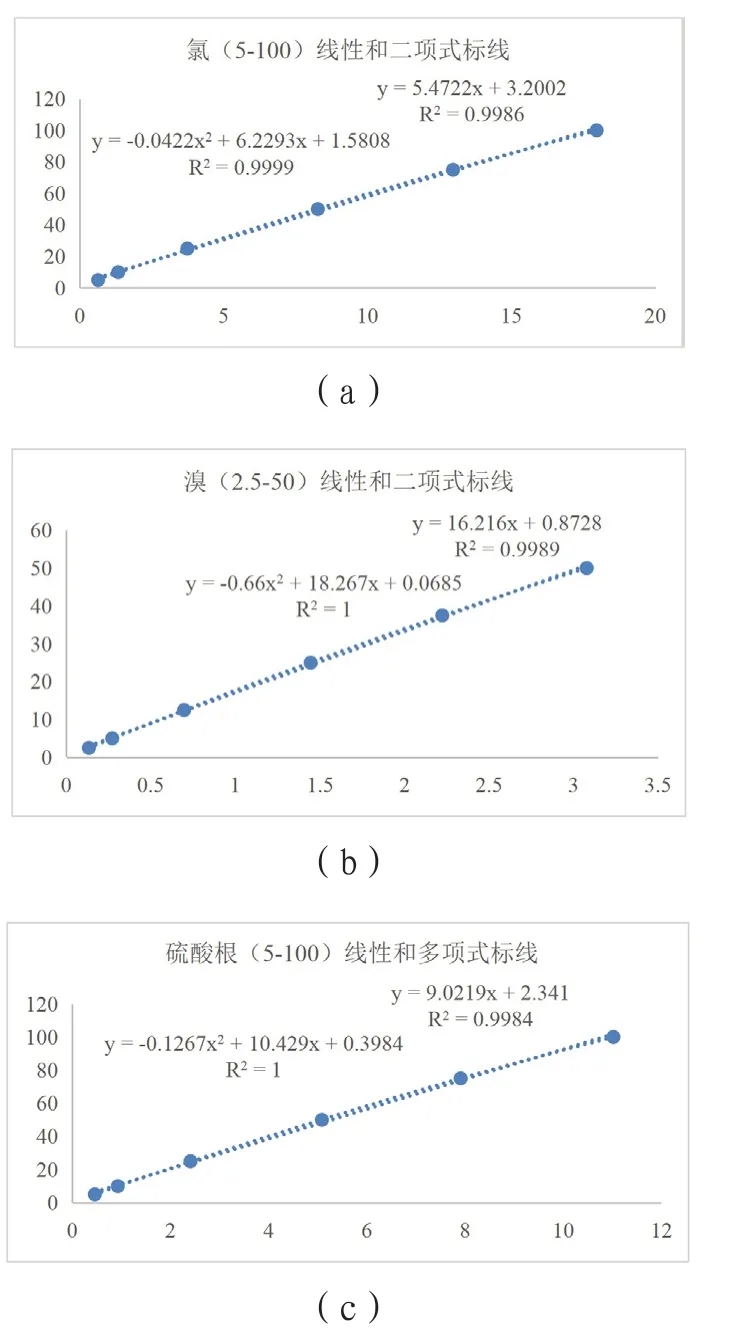
图4 氯、溴和硫酸根离子在不同浓度范围的线性拟合曲线和二项式拟合曲线图
二项式标线在实验浓度范围内计算氯、溴和硫的平均回收率(分别为101.2%、100.0%和99.9%)优于线性标线(分别为105.2%、103.7%和103.6%),且二项式标线计算的氯、溴和硫的回收率标准差(分别为4.88%、0.24%和0.45%)显著小于线性标线的计算结果(分别为14.5%、9.4%和9.2%)。说明分析试样离子浓度超出检测器线性范围时,二次拟合线更适合作为标线。因而,当试样离子浓度在检测器线性浓度范围,使用线性标线计算分析结果,否则使用二次拟合线作为标准曲线,能有效增大元素含量分析范围,提高方法的通用性。
(4)分散相微粒干扰抑制。氧弹燃烧有机危废,随后配制的水溶液试样易形成分散体系,其中微粒可能吸附被测离子化合物导致分析结果偏低,采用操作简便的加热方法有利于解吸被微粒吸附的离子化合物,从而抑制干扰。实施了一组加热(操作见2.3节)和不加热试样溶液的模拟危废氧弹燃烧-离子色谱分析。
加热处理试样溶液后的各离子空白加标回收率均提高,同时,各离子分析结果相对标准差(RSD)均减小,说明加热方法能较大程度抑制系统中微粒干扰,有利于提升分析结果的正确度和精度。
(5)方法重复性。氧弹燃烧-离子色谱分析表2中三种不同热值实际危废,考察其元素分析结果重复性。
三种不同热值危废氟、氯、溴和硫的分析结果相对标准差1.26%~4.98%,平均相对标准偏差分别为3.11%、2.13%,溴2.23%和3.03%,分析方法重复性良好。
(6)方法加标回收率。实际危废各元素加标回收率重复性良好,氟、氯、溴和硫平均回收率分别为~81%、~92%、~93%和~93%,除氟外的结果可靠。尽管氟回收率偏低,但分析结果重复性良好,其分析结果仍有参考和指导价值。
(7)方法检出限。液体石蜡中加入适量起始混标液(见2.4.2节)和乳化剂,高速搅拌制备成已知元素含量的模拟危废乳液。氧弹燃烧-离子色谱分析模拟危废,同时做空白。按下式计算信噪比S/N:

以各离子S/N不小于3:1为判据,对应元素含量确定为方法检出限。氟、氯、溴和硫含量分别为0.0400007mg/g、0.0700007mg/g、0.0400007mg/g和0.006676mg/g时,各元素的信噪比均大于3:1,所建氧弹燃烧-离子色谱法分析各元素的方法检出限小于0.047mg/g、0.077mg/g、0.047mg/g和0.007mg/g。
4.结论
氧弹燃烧量热-离子色谱分析危废过程中,采用加热法处理燃烧危废的试样溶液,有效抑制干扰离子色谱分析的不利因素,实现氧弹燃烧量热与离子色谱分析耦合。同时结合二次项标线应用,扩展离子色谱分析的元素含量范围,使得危废的氧弹燃烧量热操作参数能够与离子色谱良好匹配,实现一次同时完成危废热值与氟、氯、溴和硫分析。方法学研究表明所建方法的热值和元素分析结果重复性和回收率良好,检测灵敏度高。方法不仅通用性好,适应各类危废分析,且因无需复杂处理过程,操作简便易控,分析结果可靠。
