1株溶血色杆菌的分离鉴定与基因组分析
2022-06-24王茜月管飘萍陈诗涵黄紫贝郭鑫赵以恒罗健雅王欣宇崔颢月刘金彪王海燕刘文博
王茜月 管飘萍 陈诗涵 黄紫贝 郭鑫 赵以恒 罗健雅 王欣宇 崔颢月 刘金彪 王海燕 刘文博



摘要:从池塘中分离、纯化了1株具有极强溶血活性的革兰氏阴性杆菌BB2,在TSA培养基上菌落呈灰色、半透明圆形,经16S rRNA基因序列分析确定为色杆菌属,但无法确定到种;进一步全基因组测序结果表明,该菌株与GenBank中唯一的1株稻根色杆菌基因组同源性最高,但生物学特性又不一致。文献逆索发现其命名为稻根色杆菌主要是由于其分离于水稻根部,这对分类造成了困惑。最终依据细菌形态特征、生理生化特性、16S rRNA和全基因组序列分析及文献分析等综合信息,确定分离菌BB2为溶血色杆菌,这提醒研究者们在细菌鉴定时应谨慎使用GenBank数据序列数据,一定要对相关参考文献进行全面认真研读,获取必要的证据,为最终分类提供科学依据。本研究也为进一步研究该分离株的生物学特性和应用提供了参考和基础。
关键词:池塘;溶血色杆菌;全基因组序列;比较基因组分析
中图分类号:S182 文献标志码: A
文章编号:1002-1302(2022)12-0042-09
收稿日期:2021-08-16
基金项目:江苏高校优势学科建设工程(2018)。
作者简介:王茜月(1999—),硕士,江苏泰州人,主要从事传染病流行病学研究。E-mail:Bioyusy@outlook.com。
通信作者:刘文博(1975—),男,博士,副教授,主要从事传染病防控。E-mail:lwb@yzu.edu.cn。
溶血色杆菌,属于色杆菌属(Chromobacterium),革兰氏染色阴性,可以在多种类型的琼脂平板上生长(羊血琼脂、巧克力平板培养基、胰酪大豆胨琼脂培养基、木炭酵母提取物培养基),菌落呈灰色、圆形。在血琼脂平板上呈现出大约5 mm的明显溶血区[1-3]。目前,色杆菌属共有14个成员,分别是稻根色杆菌[4]、链烷烃色杆菌[5]、亚马逊色杆菌 [6]、水生色杆菌 [7]、氟色杆菌[8]、紫色色杆菌 [9]、鱼腥草色杆菌 [9]、伪紫色色杆菌[9]、溶血色杆菌 [1]、沼泽色杆菌 [10]、睑炎色杆菌 [10]、痘苗色杆菌 [11]、球形色杆菌 [11]和枯草色杆菌 [12]。其中,紫色色杆菌和溶血色杆菌因可以引起哺乳动物感染而受到重视[ 13-14]。由于溶血色杆菌的一些分离株能够感染人类,引起坏死性筋膜炎[1]、直肠结肠炎[15]和肺炎[3],故有必要对该菌进行更深入的研究。溶血色杆菌对β-内酰胺类抗生素有较大的耐药性[ 15]。有报道称,温带地区的河水是人感染溶血色杆菌的主要来源[2]。
16S rRNA基因因其保守性和稳定性,成为微生物分类鉴定中的重要分子标记[16],但这种方法在区分相似性很高的菌种时有局限性,全基因组分析的方法或许可以克服这一缺点[17-18],高通量测序技术的发展为微生物分类提供了新的方法和思路。平均核苷酸同源性方法具有方便、错误率低、分辨率高的特点,该方法通过计算2个基因组之间同源基因的相似性,认为ANI值接近或高于95%属于同一菌种[19]。同时,有研究表明,ANI与DDH(一般认为DDH值接近或高于70%属于同一菌种)具有较好的一致性。然而,由于成本等问题基因序列库中全基因组数据比较少,可能会由于比对时数据缺少而造成分类错误。
本研究从池塘水样中分离出1株溶血活性很强的细菌,确定为色杆菌属,但在确定其种时却因为数据、文献问题造成了困惑,最终综合其16S rRNA序列、全基因组序列、生物学特性和文献综合分析,确定为溶血色杆菌,现报告如下。
1 材料与方法
1.1 材料
1.1.1 主要仪器 生物安全柜购自Thermo公司;电子天平购自上海精密仪器仪表有限公司;奥林巴斯显微镜购自日本奥林巴斯公司;高压灭菌锅购自Tomy公司;PCR仪2720购自美国ABI公司;电泳仪和凝胶成像系统购自美国伯乐公司。
1.1.2 主要试剂 血平板购自江门市凯林贸易有限公司;TSA琼脂购自青岛海博生物技术有限公司;16s rDNA Bacterial Identification PCR Kit购自宝生物工程(大连)有限公司。
1.2 细菌分离、纯化、16S rRNA 基因扩增
2019年8月,采集扬州大学文汇路校区农学大楼和兽医学院大楼中间池塘内的水样,4 000 r/min离心后取上清液,接种于含有10%血清的血平板中,于37 ℃条件下培养24 h。挑取单个菌落,接种血平板、TSA等进行菌落特性研究,连续纯化3代,对单个菌落进行革兰氏染色、镜检,扩增16S rRNA基因,送往擎科生物技术公司测序。
1.3 16S rRNA 基因序列比对
基于EZBioCloud 16S数据库(https://www.ezbiocloud.net/),对细菌进行 16S rRNA基因比对,使用MEGA-X软件,构建Neighbor-Joining系统进化树进行遗传进化分析。
1.4 基因组DNA的提取
挑取单个菌落,提取高质量基因组DNA,利用Nanodrop、Quibit和 0.35% 琼脂糖凝胶电泳验证后,送武汉贝纳科技服务公司测序。
1.5 基因组测序和组装
对Nanopore測序仪测得的原始数据进行过滤,去除接头、短片段及低质量数据。使用unicycler (0.4.8)[20]软件对过滤后的reads进行组装。使用bwa[21]软件将二代测序illumina短序列比对到拼接完成的基因组上,使用minimap2将3代测序的长序列比对到拼接完成的基因组上,使用samtools depth工具,以2 000 bp为一滑窗在基因组上滑动,统计平均测序深度。通过Prokka软件对组装后的基因组进行编码基因预测。利用预测得到的基因组信息,如基因组测序深度、GC分布及基因组结构注释进行整合,用R包circlize绘制基因组圈图。将全基因组序列信息上传NCBI数据库,GenBank登录号为CP061849。
1.6 基因组成分初步分析
通过PHAST软件来预测前噬菌体基因[22];通过RepeatMasker软件对基因组进行重复序列预测[23];通过IslandView4网站预测基因组中存在的基因岛[24]。
1.7 抗性基因预测
通过Comprehensive Antibiotic Resistance Database(CARD)[25]数据库对细菌的抗性基因进行筛查,获得基因的抗性类型、抗性谱等详细信息,预测细菌的耐药基因。
1.8 比较基因组分析
1.8.1 全基因组序列比较及共线性分析 从GenBank数据库中查找色杆菌属可获得全基因组序列的菌株,包括稻根色杆菌(C. rhizoryzae JP2-74)、溶血色杆菌(C. haemolyticum CH06-BL)、紫色色杆菌(C. violaceum ATCC 12472)、痘苗色杆菌(C. vaccinia 1-1)的基因组信息,与分离菌的基因组序列直接进行比较分析,统计基因组基本特征,计算平均核苷酸同源性(ANI)及DNA-DNA杂交值(DDH)。采用Mauve软件,构建参考菌株和分离菌的全基因组序列共线性比对[26]。
1.8.2 基因家族比较分析 利用OrthoVenn2在线工具[27](https://orthovenn2.bioinfotoolkits.net/)对分离菌和“1.8.1”节所选参考菌株的蛋白序列进行直系同源聚类分析(参数:e-value≤1×10-5,Inflation value≤1.5)。
2 结果与分析
2.1 细菌分离、纯化和16S rRNA 基因扩增
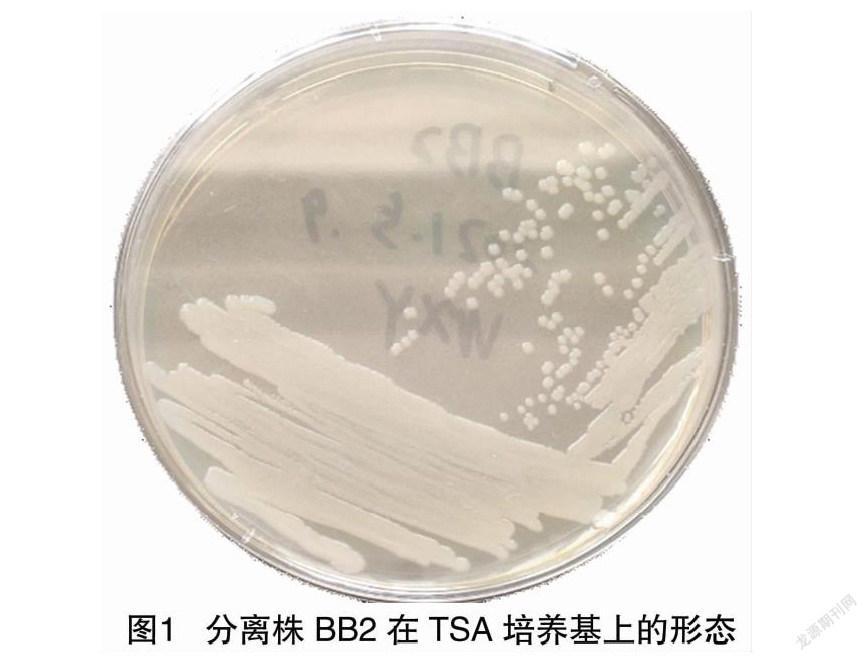
从池塘水样中划线分离培养出不同菌落形态的多种细菌,其中有1种细菌在TSA培养基上呈灰色、半透明的圆形菌落(图1),多次纯化后,该菌可以在血琼脂平板上产生非常明显的溶血环(图2),符合溶血色杆菌的形态学特点和溶血特性。16S rRNA 基因扩增成功。
2.2 16S rRNA 基因序列比对结果
通过扩增细菌的16S rRNA基因并测序得到其序列,进行BLAST比对,结果表明为色杆菌属,下载相关的16S rRNA基因进行比对分析,结果发现该细菌的16S rRNA基因序列与Chromobacterium haemolyticum MDA0585、Chromobacterium rhizoryzae LAM1188、Chromobacterium alkanivorans IITR-71、Chromobacterium aquaticum CC-SEYA-1较为接近,相似度分别为99.38%、99.30%、99.09%、98.56%。将分离到的细菌命名为BB2,遗传进化树见图3。
2.3 测序结果统计并绘制基因组圈图
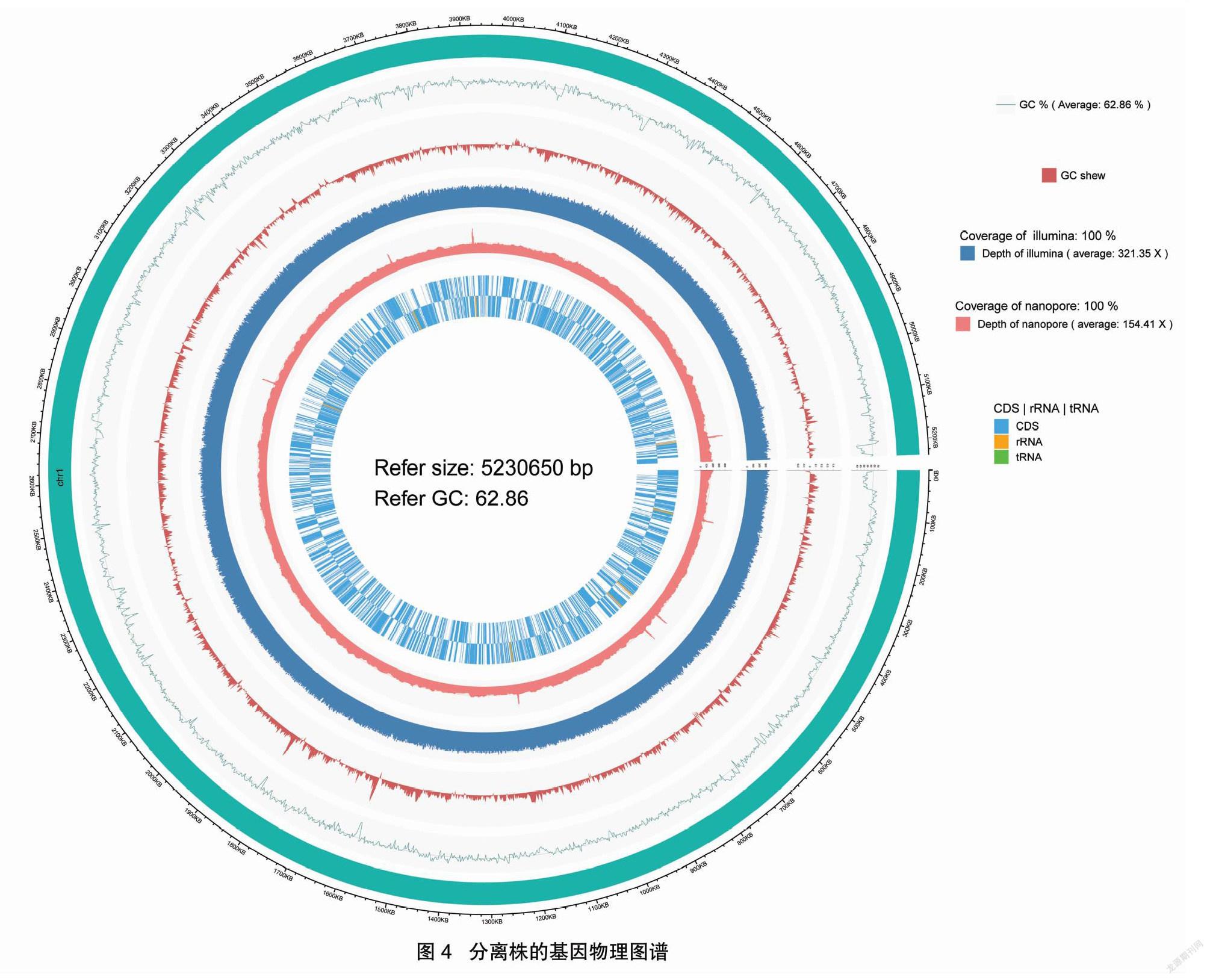
高通量测序仪测序得到5 635 123条序列,过滤后得到5 625 367条序列,二代测序平均测序深度为321.35 X,三代测序平均测序深度约为154.41 X。过滤后细菌基因组总长度为5 30 650,GC含量为63.09%,基因组中重复序列含量极少,总长为 59 951 bp,占基因组序列全长的1.15%。经预测,细菌基因组含有4 859个编码基因,4 690个CDS,87个tRNA,25个rRNA,1个tmRNA,绘制基因组圈如图4所示。
2.4 基因组成分初步分析
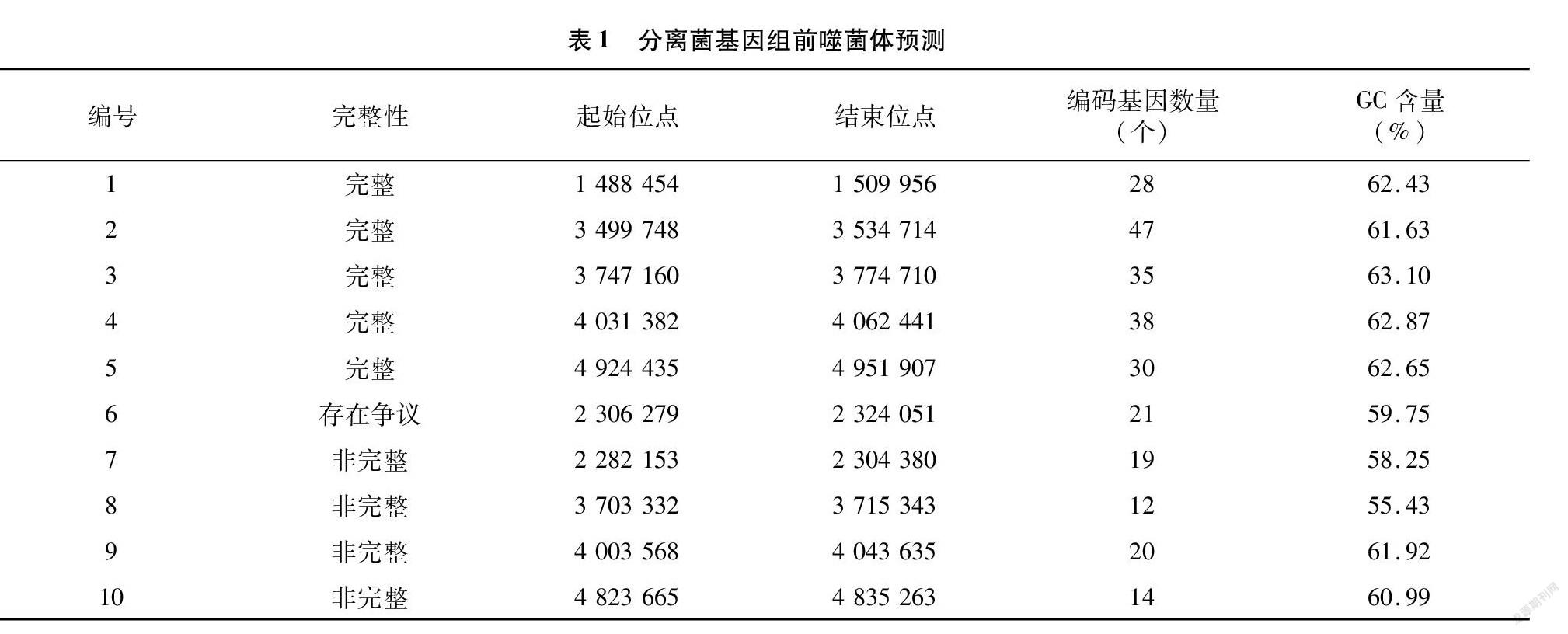
2.4.1 前噬菌体分析 PHASTER预测结果显示,分离菌中含有5个完整的前噬菌体,分离菌作为溶源性细菌,与病原菌共同培养时,前噬菌体可能会从宿主染色体上切割下来,大量复制,成为成熟的噬菌体,通过噬菌体的传染,导致病原菌裂解、死亡。分离菌还含有4个非完整的前噬菌体,分别含有19、12、20、14个CDS,可能是在此发生了基因突变,导致产生了非完整的前噬菌体。此外,分离菌还存在1个有争议的前噬菌体,含有21个CDS。具体情况见表1。
2.4.2 基因岛预测 基因岛通常被认为通过水平转移获得,这些区域常包含抗生素抗性及毒力相关基因。基因岛预测对细菌的进化分析,以及在进化过程中可能获得的特异功能的研究有重要意义。分离菌的全基因组中共预测到了37个基因岛,其中,IslandPath-DIMOD方法預测到15个基因岛,SIGI-HMM方法预测到16个基因岛,IslandPick方法预测到6个基因岛,这些基因岛可能对细菌适应不同的环境和抗生素耐药性有重要作用。预测结果见图5。
2.5 抗性基因预测结果
根据CARD抗性基因数据库预测结果,该菌株含有adeF外排泵和抗生素靶点改变机制(抗磺胺类药物)。
2.6 比较基因组分析
2.6.1 全基因组序列比较分析 由表2可见,分离菌的基因组大小和GC含量与C.rhizoryzae JP2-74和C.haemolyticum CH06-B较为接近,C. violaceum ATCC 12472与其他菌株基因组基本特征差异较大。一般认为ANI值接近或高于95%划分为同一菌种[16],令人惊讶的是,按照此标准,分离菌和C. rhizoryzae JP2-74及C.haemolyticum CH06-BL均属于同一菌种,而和C. violaceum ATCC 12472及C. vaccinia 1-1显然不属于同一菌种。DDH预测值认为物种边界的阈值为70%左右,高于70%被认为属于同一菌种[19],DDH值预测结果与ANI值的结果具有一致性。
通过共线性分析可以进一步识别菌种全基因组的一致性和变异性,体现基因组的共同起源。分离菌与4株参考菌株的共线性分析结果见图6。由图6可见,分离菌与C. rhizoryzae JP2-74、C. haemolyticum CH06-BL存在大量同源性区域,但也存在缺失、插入和重排区域,与C. violaceum ATCC 12472、C. vaccinia 1-1差异较大。
2.6.2 直系同源聚类分析 OrthoVenn2 直系同源聚类分析结果显示,共得到5 324个基因簇,2 462个同源基因簇(至少包含2个物种)以及2 862个单拷贝基因簇。由图7可以看出,5株菌的共有基因簇(核心基因簇)有2 898个,分离菌和C. rhizoryzae JP2-74及C. haemolyticum CH06-BL的共有基因簇较多,和C. vaccinia 1-1的共有基因簇较少。相似矩阵热图(图8)也显示,分离菌和C. rhizoryzae JP2-74及C. haemolyticum CH06-BL具有较高的聚类。
以上结果均表明全基因组分类方法可以区分溶血色杆菌、紫色色杆菌和痘苗色杆菌。虽然溶血色杆菌和稻根色杆菌在基因组的组成上极为相似,单靠全基因组测序分析方法仍无法做出判断,但是由于稻根色杆菌目前仅有从水稻根部分离出的报道[4],且基因库中相关的全基因组序列很少,进一步进行文献综合分析,结合分离菌的细菌生物学特征和极强的溶血特性,认为BB2应该属于溶血色杆菌。
3 讨论
通过16S rRNA基因序列和遗传关系鉴定细菌、确定菌种已成为常用的细菌分类手段,但其分类仅仅能够到属,无法确定到种,现在很多研究者对该方法的原理并不清楚,在GenBank进行BLAST,同源性最高的就确认到种,这种文章现在越来越多,但并未深究其因。然而,GenBank中的数据均为测定者上传,全面性和准确性值得商榷,且其分类依据也是依赖于以往的基因库,这就造成了以讹传讹的后果。16S rRNA序列作为细菌分类的依据,一般分别以97%和95%作为划分一种新种和新属的依据[28],2006年划分新种的标准被提高到了98.7%[29],这一标准和DDH结果有较好的一致性[30]。但是仅仅依赖于16S rRNA基因序列不能够确定到种,只能作为参考。本研究中细菌纯化后测定的序列表明分离株BB2为色杆菌属。基于EZBioCloud 16S 数据库(https://www.ezbiocloud.net/)对该分离株的16S rRNA基因进行比对以鉴定菌种,结果发现该细菌的16S rRNA基因序列与Chromobacterium haemolyticum MDA0585、Chromobacterium rhizoryzae LAM1188、Chromobacterium alkanivorans IITR-71、Chromobacterium aquaticum CC-SEYA-1较为接近,相似度分别为99.38%、99.30%、99.09%、98.56%。确定分离菌属于色杆菌属,但无法确定菌种,这也体现出了16S rRNA序列比对在区分同属不同种细菌之间的局限性。
随后的全基因组序列测定结果与GenBank中可获得的色杆菌属成员进行基因组比较分析,结果显示,分离菌和唯一的1株稻根色杆菌Chromobacterium rhizoryzae LAM1188基因组特征最相似,遗传关系很近。但是很多参考菌株并没有测定全基因组序列,缺少大量的序列证据,无法对其进行种的确定,进一步回顾将稻根色杆菌确定为新种的报道[4],发现其依据主要是表型、DNA-DNA杂交值和16S rRNA序列比对,其中,报道中用来比较的2株模式菌株稻根色杆菌C. rhizoryzae LAM1188和溶血色杆菌C. haemolyticum MDA0585 16S rRNA序列相似度为98.7%,这个值恰好是确定细菌为新种的边界值,这说明16S rRNA序列比对在本研究中确实不能作为鉴定菌种的证据。该报道最终是以表型和DNA-DNA杂交试验来确定菌种的,稻根色杆菌在TSA培养基上呈现黄褐色,但溶血色杆菌在TSA培养基上呈现灰色。本研究中分离菌BB2在TSA培养基上也呈现灰色,故最终确定BB2为溶血色杆菌。
在平常的菌种鉴定过程中,依据GenBank中的序列进行细菌分类,主要包括16S rRNA序列比对和基因组分析方法。但在本研究中,如果仅仅依据16S rRNA序列比对和基因组分析方法,很有可能会得出分离菌BB2属于稻根色杆菌的错误结论。造成这种情况的原因可能有2点,一是稻根色杆菌和溶血色杆菌基因組组成极其相近;二是数据库中登记信息有误,实际上这株稻根色杆菌应是1株溶血色杆菌。因为稻根色杆菌的模式菌株基因组序列未见上传,且由于生物材料缺乏,也无法进行 DNA-DNA 杂交试验验证,故具体原因仍待进一步探究。这提醒研究者们如果完全依据GenBank上的序列数据进行分类,不去考虑表型和生理生化特性,会造成偏差和错误,应依据细菌形态特征、生理生化特性、16S rRNA和全基因组等综合分类,有困惑时一定要对相关参考文献进行全面认真研读,获取必要的证据,做出准确判断。已知溶血色杆菌能够感染人类,稻根色杆菌仅有1篇报道,未有感染性报道。本研究为谨慎使用GenBank数据和文献提供了一些证据,也为进一步研究该溶血色杆菌分离株提供了参考和基础。
参考文献:
[1]Han X Y,Han F S,Segal J. Chromobacterium haemolyticum sp. nov.,a strongly haemolytic species[J]. International Journal of Systematic and Evolutionary Microbiology,2008,58(6):1398-1403.
[2]Kanamori H,Aoyagi T,Kuroda M,et al. Chromobacterium haemolyticum pneumonia associated with near-drowning and river water,Japan[J]. Emerging Infectious Diseases,2020,26(9):2186-2189.
[3]Takenaka R,Nureki S I,Ueno T,et al. Chromobacterium haemolyticum pneumonia possibly due to the aspiration of runoff water[J]. Japanese Journal of Infectious Diseases,2015,68(6):526-529.
[4]Zhou S,Guo X,Wang H M,et al. Chromobacterium rhizoryzae sp. nov.,isolated from rice roots[J]. International Journal of Systematic and Evolutionary Microbiology,2016,66(10):3890-3896.
[5]Bajaj A,Kumar A,Yadav S,et al. Isolation and characterization of a novel Gram-negative bacterium Chromobacterium alkanivorans sp. nov.,strain IITR-71T degrading halogenated alkanes[J]. International Journal of Systematic and Evolutionary Microbiology,2016,66(12):5228-5235.
[6]Menezes C B A,Tonin M F,Corrêa D B A,et al. Chromobacterium amazonense sp. nov. isolated from water samples from the Rio Negro,Amazon,Brazil[J]. Antonie Van Leeuwenhoek,2015,107(4):1057-1063.
[7]Young C C,Arun A B,Lai W A,et al. Chromobacterium aquaticum sp. nov.,isolated from spring water samples[J]. International Journal of Systematic and Evolutionary Microbiology,2008,58(4):877-880.
[8]Moss M O,Ryall C,Logan N A. The classification and characterization of chromobacteria from a lowland river[J]. Journal of General Microbiology,1978,105(1):11-21.
[9]Kmpfer P,Glaeser S P,Soby S D. Chromobacterium pseudoviolaceum is a later heterotypic synonym of Chromobacterium violaceum Bergonzini 1880[J]. International Journal of Systematic and Evolutionary Microbiology,2018,68(9):2967-2968.
[10]Blackburn M B,Farrar R R Jr,Sparks M E,et al. Chromobacterium paludis sp. nov.,a novel bacterium isolated from a Chesapeake Bay marsh[J]. International Journal of Systematic and Evolutionary Microbiology,2020,70(12):6142-6146.
[11]Soby S D,Gadagkar S R,Contreras C,et al. Chromobacterium vaccinii sp. nov.,isolated from native and cultivated cranberry (Vaccinium macrocarpon Ait.) bogs and irrigation ponds[J]. International Journal of Systematic and Evolutionary Microbiology,2013,63(Pt 5):1840-1846.
[12]Martin P A W,Gundersen-Rindal D,Blackburn M,et al. Chromobacterium subtsugae sp. nov.,a betaproteobacterium toxic to Colorado potato beetle and other insect pests[J]. International Journal of Systematic and Evolutionary Microbiology,2007,57(5):993-999.
[13]修云芳,邵良平,李碧春,等. 紫色色桿菌感染小熊猫引起肺炎的临床调查[J]. 兽类学报,201 31(1):108-112.
[14]Okada M,Inokuchi R,Shinohara K,et al. Chromobacterium haemolyticum-induced bacteremia in a healthy young man[J]. BMC Infectious Diseases,2013,13:406.
[15]Tanpowpong P,Charoenmuang R,Apiwattanakul N. First pediatric case of Chromobacterium haemolyticum causing proctocolitis[J]. Pediatrics International,2014,56(4):615-617.
[16]Ramasamy D,Mishra A K,Lagier J C,et al. A polyphasic strategy incorporating genomic data for the taxonomic description of novel bacterial species[J]. International Journal of Systematic and Evolutionary Microbiology,2014,64(Pt ):384-391.
[17]Tindall B J,Rosselló-Móra R,Busse H J,et al. Notes on the characterization of prokaryote strains for taxonomic purposes[J]. International Journal of Systematic and Evolutionary Microbiology,2010,60(Pt 1):249-266.
[18]Chun J,Oren A,Ventosa A,et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes[J]. International Journal of Systematic and Evolutionary Microbiology,2018,68(1):461-466.
[19]Meier-Kolthoff J P,Auch A F,Klenk H P,et al. Genome sequence-based species delimitation with confidence intervals and improved distance functions[J]. BMC Bioinformatics,2013,14:60.
[20]Wick R R,Judd L M,Gorrie C L,et al. Unicycler:Resolving bacterial genome assemblies from short and long sequencing reads[J]. PLoS Computational Biology,2017,13(6):e1005595.
[21]Li H,Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform[J]. Bioinformatics,2010,26(5):589-595.
[22]Zhou Y,Liang Y J,Lynch K H,et al. PHAST:a fast phage search tool[J]. Nucleic Acids Research,201 39(Web Server issue):347-352.
[23]Tarailo-Graovac M,Chen N S. Using repeatmasker to identify repetitive elements in genomic sequences[M]//Current protocols in bioinformatics. New York:John Wiley & Sons,2009.
[24]Bertelli C,Laird M R,Williams K P,et al. IslandViewer 4:expanded prediction of genomic Islands for larger-scale datasets[J]. Nucleic Acids Research,2017,45(W1):30-35.
[25]Alcock B P,Raphenya A R,Lau T T Y,et al. CARD 020:antibiotic resistome surveillance with the comprehensive antibiotic resistance database[J]. Nucleic Acids Research,2019,48(D1):517-525.
[26]Darling A C E,Mau B,Blattner F R,et al. Mauve:multiple alignment of conserved genomic sequence with rearrangements[J]. Genome Research,2004,14(7):1394-1403.
[27]Xu L,Dong Z B,Fang L,et al. OrthoVenn2:a web server for whole-genome comparison and annotation of orthologous clusters across multiple species[J]. Nucleic Acids Research,2019,47(W1):52-58.
[28]Stackebrandt E,Frederiksen W,Garrity G M,et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology[J]. International Journal of Systematic and Evolutionary Microbiology,2002,52(Pt 3):1043-1047.
[29]Stackebrandt E,Ebers J. Taxonomic parameters revisited:Tarnished gold standards[J]. Microbiology Today,2006,8(4):6-9.
[30]Rosselló-Mora R. DNA-DNA reassociation methods applied to microbial taxonomy and their critical evaluation molecular identification,systematics,and population structure of prokaryotes[M]. Berlin:Springer,2006:23-50.
