海水中腐殖质对溶解态铁的络合及其影响因素
2022-06-22姚佳佳杨茹君张莹莹
姚佳佳, 吴 瑶, 杨茹君, 张莹莹
(1. 中国海洋大学 化学化工学院, 山东 青岛 266100; 2. 盐城工学院 环境科学与工程学院江苏省环境保护海涂生态与污染防治重点实验室, 江苏 盐城 224051)
腐殖质(humic substances, 简称HS)是地球表面上天然有机物最广泛的存在形式, 大约占溶解有机物(dissolved organic matter, DOM)的15%~80%[1-2], 是其主要组成部分。腐殖质是一种复杂的混合物, 由死亡的植物和微生物通过复杂的生物和化学过程形成[3-4]。根据水溶性可将其分为两类, 一类为腐殖酸(humic acid,简称HA, 或称为胡敏酸)可溶于碱性溶液, 但不溶于强酸溶液。另一类为富里酸(fulvic acid, 简称FA, 或称为黄腐酸), 可溶于酸和碱[5]。由于腐殖质化学组成复杂, 并且含有羟基、羧基、羰基等多种含氧官能团[6], 对金属离子(包括Fe, Mn, Cu和Zn等)具有螯合能力和高的亲和力。因此能够通过络合和共沉淀与土壤和水生环境中的各种金属离子发生强烈的相互作用, 进而影响金属离子的浓度、生物可利用性等[7-11]。例如腐殖质可与土壤中的重金属络合[8,12], 通过改变重金属的存在形态降低其生物毒性, 提高微生物活性。海水中痕量金属如Cu、Pb和Fe的浓度极低, 所以HS的络合作用会直接影响其浓度、迁移和转化, 特别是对于溶解态Fe[13-14]。此外, 陆源HS广泛分布于淡水、河口和沿海水域, 且浓度较高[14-15], 在控制河口和沿岸水体中金属离子的迁移和转化等生物地球化学行为方面起着主导作用。本文介绍了HS的种类、来源和分布及对海水中溶解态Fe迁移转化的影响, 包括溶解态Fe分布、生物可利用性和氧化还原转化等。
1 水生环境中腐殖质的种类、来源及分布
腐殖质是由疏水性、难降解性和多相性的有机物组成的非均匀混合物[4], 正是这种独特的异质性使其元素组成、化学功能和分子大小分布复杂多样[16]。除了能够与金属离子发生相互作用外, 腐殖质还具有许多环境功能, 包括缓冲营养物质浓度、参与碳循环以及调节光驱动反应等[17]。目前仪器检测到的海水中HS通常是HA和FA的混合物, 将HS进一步提纯、分离, 可得到纯化的HA和FA。尽管两者都属于腐殖质, 但官能团数量的不同[17-18]导致其理化性质上存在很大差别。FA的平均浓度大约是HA的两倍[13,19], FA是河流和河口水域中HS的主要组成部分, 而HA仅占一小部分(~10%)[20]。除溶解度比HA更大外, FA的分子量(1~2 KDa)通常明显低于HA(4~200 KDa)[17], 在河口混合的过程中, 溶解度小的HA在早期更容易通过絮凝和沉淀等过程去除,而FA则不易产生絮凝和沉淀[20-21]。此外FA中相对难降解的木质素酚含量高[22], 因此涉及FA的降解过程比较缓慢。
腐殖质一般由植物体经微生物分解产生, 由于陆地和海洋中的生物种类不同, 由此降解后的腐殖质也存在差别, 分别称为陆源或海源腐殖质。陆源腐殖质是木质素或其他高等植物的降解产物[23-24], 具有较高的芳香性, 可通过降雨进入河流中, 并输入河口、近海[25]; 海源腐殖质与浮游生物及微生物活动密切相关[26], 富含羧基等脂肪性较高的基团。沿海水域的HS主要来源于陆地, 而开放海洋的HS主要来源于海洋。受到陆源HS输入的影响, 西北大西洋海岸中腐殖质的质量浓度高达189 μg/L, 而在开放海洋中的质量浓度较低, 如大西洋为12~116 μg/L,太平洋为18~54 μg/L, 南大洋为18~81 μg/L, 并且陆源腐殖质只占其溶解腐殖质库中的很小一部分[2]。
HS在水生系统中之所以被认为是难降解的,是因为其具有的复杂分子结构能抵抗微生物的攻击。但是有研究表明它们也可能被微生物分解, 并通过各种物理化学过程去除[27]。例如陆源腐殖质在河口和沿岸水域受到重要的细菌降解[28], 而光化学降解容易发生在河流运输过程中[29]。由于水体中HS主要以胶体形式存在[30], 容易受到离子强度变化的影响, 特别是当河口淡水与海水混合时, 大量带正电荷的离子使HS趋于絮凝。Fox等[31]的早期研究就已经表明HS的胶体性质使其在电解质溶液和海水中会迅速絮凝。因此它们在河流、湖泊和河口的浓度相对较高, 达到几个mg/L, 而在开放海洋浓度仅为数十至数百 μg/L[32]。在垂直分布上,Gledhill等[33]的结果发现腐殖质类FDOM在大西洋表层海水中的浓度相对较低, 随深度增加而增加,可能是由于有机质产生的海洋腐殖质在深海积累。与此相反, Slagter等[34]和Hassler等[35]发现北冰洋和太平洋HS浓度从表层向深海降低, 表明水柱中可能存在HS的矿化。
2 海水中HS对溶解态铁迁移转化的影响
铁(Fe)是海洋中浮游植物的生长所必需的微量元素, 其在海水中的极低溶解度(~10–11mol/L)限制了40%海洋的初级生产[36-37]。有机配体是影响Fe溶解度、生物可利用性的主要因素, 通过与Fe的络合不仅能够增大海水中Fe的溶解度, 而且还能阻止Fe的清除如水解、沉淀, 从而增加其生物可利用性, 进而促进Fe的长距离迁移和转化[38]。有机络合的强度与有机配体的种类及来源密切相关。在海洋环境中,铁载体及其降解产物、糖类和腐殖质都是潜在的铁结合有机配体, 而腐殖质是天然水体中有机配体的主要组成部分[13]。水体中的HS具有较强的金属结合能力, 可以与不同的金属离子发生络合作用[39-40]。与此同时, 各金属离子与HS的络合之间也会存在竞争关系[41-42]。对稳定常数的研究表明, 不同金属HS络合物的稳定性顺序一般符合Irving-Williams序列,即Mg
2.1 海水中HS对溶解态铁分布的影响
在已有关于河口[15,46]、沿岸[11,47]和开放海洋[48]的研究发现HS与溶解态Fe(dissolved iron, DFe)浓度变化都显示出高度的相关性, 表明HS控制着Fe的生物地球化学行为。由于HS为胶体结构, 在河口混合区域, 随着盐度的增加, 电解质浓度逐渐增大, HS发生絮凝而被清除, 导致溶解态铁也同时被沉降。河口系统的早期经典研究发现Fe与HS的共沉淀导致Fe在河口区的去除率达到90%以上, 其浓度从0.5~10 μmol/L[49]降低到1~20 nmol/L[13,49], 并且呈沿海(2.8~2.9 nmol/L)到大洋(0.24~0.27 nmol/L)逐渐降低的趋势, Sholkovitz等[50]的进一步研究表明大多数损失的溶解态Fe是胶体态部分。Yang等[15]测定了长江口在盐度梯度(0.14~33)下DFe、FA和HA的浓度变化, 发现随着盐度的升高三者浓度都呈现相似的降低趋势, 其去除率分别高达96%、74%和67%, 并且盐度与DFe、FA和HA浓度之间的关系可以用一阶指数去除模型来描述。在该研究中HA和FA的去除是由于淡水与海水混合过程中的絮凝作用, 而FA和HA沿盐度梯度的絮凝则导致了DFe的去除。Mahmood等[51]在默西河河口也发现了类似的现象:盐度最低时DFe和HS浓度最高, HS随盐度的增加表现出非保守性, 在盐度范围为16.38~34.18两者发生共沉淀导致DFe的损失。在米利卡河口[52]和叶尼塞河河口[53]DFe的去除率分别达到了96%和98%,河口沉积物中HS的絮凝作用也是去除溶解铁的主要原因。由此可以看出, HS对河口混合过程中铁的去除起着重要作用。
除了去除作用外, 河流中陆源HS的络合使DFe迁移至开放海洋, 为海洋中浮游植物的生长提供了更多的生物可利用铁。Krachler等[54]的研究证明流经泥炭地的河流中含有高浓度的HA和FA等有机配体, 为海洋提供了3 300 nmol/L DFe, 是世界平均河流(~40 nmol/L)的近100倍。泥炭地河流是日本津加鲁海峡沿岸水域中DFe最重要的来源[55],预计每年为全球海洋贡献1.5×1010mol的DFe[56]。Krachler等[25]也发现陆源HS浓度随着离岸距离的增加而迅速降低, 并且通过放射性同位素示踪法发现陆源HS增大了海水中Fe(Ⅲ)的溶解度。Yamashita等[57]报道了在鄂霍次克海浅层沉积物中陆源腐殖质携带溶解铁向北太平洋迁移。爱尔兰海河口和沿岸水域中DFe和陆源HS变化的一致性也表明了陆源HS对Fe迁移的控制[13]。因此, 腐殖质对海洋生物可利用铁的供应具有重要的作用, 特别是陆源腐殖质大大增加了沿岸水域溶解铁的浓度并支持浮游植物的生长, Fe-HS络合物的稳定性也使铁从河口运输到开放海洋。
HS在沿海水域中大量存在, 在深海中也以低浓度存在, 因此HS与DFe的络合不仅在河口、沿海等水体中很重要, 对海水中溶解铁的贡献也不可忽略[54,56]。Laglera等[32]发现在富含HS的北极表层水域中有49%的HS与Fe发生络合, 且Fe-HS配合物对DFe浓度的总体贡献高达80%, 高浓度的Fe-HS络合物使得北冰洋表层水中的DFe能够跨极地漂流。地中海地区DFe和HS也存在相似的分布,两者浓度具有显著相关性(R2>0.58;P<0.05)[48], 斜率即DFe/eHS率为13±2.5 nmol DFe/mg eq. SRFA,表明HS通过络合DFe参与铁的循环。Bundy等[47]对旧金山湾中的DFe、配体和HS进行分析测定, 发现HS浓度与配体浓度存在直接的线性关系(R2=0.95,P<0.05), 同时, HS还与溶解态Fe存在某种线性关系, 这表明HS可能是配体的主要组成部分,对Fe的分布起到重要作用。Nishimura等[58]通过测定HS的荧光强度来研究HS与DFe的关系, 结果表明在加拿大西北部育空河口区, 腐殖质类FDOM与DFe浓度成正相关, 并且对于DFe向白令海陆架区的运输起到重要的作用。在北太平洋的中心海域DFe与腐殖质类荧光强度表现出强线性相关, 并确定腐殖质类FCOM是深层水体中络合溶解态铁的天然有机配体, 对溶解态铁在海水中的存留具有重要意义。在鄂霍次克海、白令海以及太平洋中层和深层水中, DFe也受到腐殖质类FDOM等天然有机配体的络合作用的控制[59-60]。上述结果均表明腐殖质类FDOM可以通过提供有机配体来控制DFe的分布。因此在开阔大洋中, HS不仅能影响溶解态铁的运输如跨极迁移, 还能增大溶解态铁的溶解度并使其稳定存在。
2.2 海水中HS对溶解态铁生物可利用性的影响
HS是海洋中DFe的重要有机配体, 而有机配体是调控铁反应活性及其对海洋浮游植物生物可利用性的主要因素[61]。有机质增强浮游植物获取铁的机制主要包括2种, 一种是酶还原机制: 浮游植物细胞表面具有可诱导的还原酶, 介导有机结合Fe(Ⅲ)的还原及Fe的解离, 随后细胞表面的转运体将无机铁内化[62]。另外一种是光还原机制: 在光照条件下, 有机配体络合的 Fe(Ⅲ)会发生还原, 而Fe(Ⅱ)与配体之间较低的亲和力导致铁的释放, 促进藻类吸收[63]。因此有机络合铁的生物活性受到氧化还原和光化学过程的影响, 其中光化学在介导铁还原解离中具有重要作用, 对铁的生物可利用性不可忽略。作为铁的重要有机配体, HS也能通过络合作用影响DFe的生物可利用性, 研究表明与HS络合的Fe能够转化成更不稳定的形式, 更易于被浮游植物吸收, 因此与HA或FA结合的Fe对浮游植物都具有高度的生物可利用性[33,64-66], 尤其是在河口和近岸水中。
不同有机配体的铁结合强度与稳定性会导致生物可利用性的差异。一方面, 一些有机配体可能降低铁对浮游植物的生物可利用性如DFB[67], 另一方面, 有机络合可以增加铁在海水中的溶解度, 甚至使其生物可利用性高于无机铁。Chen等[68]使用去铁胺B(DFB)、高铁色素(FC)、HA三种不同的配体模型与Fe络合, 来研究它们对近岸硅藻的同化速率。不同Fe种初始同化速率的顺序为无机Fe≥HA-Fe(Ⅲ)>DFB-Fe(Ⅲ) ≈ FC-Fe(Ⅲ)。HA-Fe(Ⅲ)的高生物可利用性表明HS不仅能控制铁的地球化学行为, 还为海洋浮游植物提供Fe促进其生长。Krachler等[69]提取泥炭沼泽河口区水体中的HS与绿藻(Chlorella salina)一起培养, 结果发现含HS的培养瓶中藻类生长最快, 这说明来源于泥炭沼泽的HS能够提高铁对海藻的生物可利用性。Fe-HS的生物可利用性与浮游植物的种类也有一定的影响, 比如其对硅藻(Thalassiosira pseudonana)有较高的生物可利用性, 而对海洋蓝细菌(Synechococcus sp.)的生物可利用性较弱[68]。腐殖质本身也能刺激沿海浮游植物的生长, Krachler等[69]在实验室培养中发现HS的模型物对藻类的生长具有强的促进作用。Fe是浮游植物重要的营养素, HS能够增大浮游植物对铁的吸收, 浮游植物又通过释放HS进一步调节Fe的分布、化学形态和循环, 因此Fe、HS与浮游植物之间的作用是相互的。
2.3 海水中HS对溶解态铁氧化还原的影响
HS含有多种显色官能团, 能够引发许多重要的光化学反应, 是一种公认的光敏化剂。光照使HS产生溶剂化电子, 以及多种活性氧(ROS)如羟自由基(·OH)、过氧化氢自由基(ROO·)和单线态氧(1O2)、对天然水体中的金属如 Fe[6,71]及有机物如苯酚[70]的氧化还原转化具有不可忽略的作用。
Fe循环的关键特征包括Fe(Ⅲ) -配体的光解、Fe(Ⅲ)还原为Fe(Ⅱ)以及配体的氧化, 因此Fe-HS络合物影响着Fe(Ⅲ)和Fe(Ⅱ) 的氧化还原转化[72]。HS在光照作用下失去电子被氧化, 并将电子传递给Fe(Ⅲ), 使其得到电子还原为Fe(Ⅱ), 或被 2O-·还原, 而Fe(Ⅱ)的氧化主要通过与溶解氧O2、H2O2以及 2O-·自由基等发生氧化反应完成。此外, 还能通过配体-金属电荷转移(LMCT)来影响铁的光化学还原速率, 从而促进水体中浮游植物对铁的吸收, 增强全球铁循环[73]。早在19世纪80年代, Miles等[74]研究天然水体中的腐殖质和铁的氧化还原反应循环, 在这个循环中HS作为催化剂发生LMCT反应,包括将Fe(Ⅲ)光还原成Fe(Ⅱ), 随后溶解氧将Fe(Ⅱ)氧化成Fe(Ⅲ), 进而促进天然水体中的氧消耗。Voelker等[75]发现Fe(Ⅲ)与FA的络合物能够发生LMCT反应, 生成Fe(Ⅱ)。该过程主要发生在溶解氧浓度较高的表层水中, 光照激发HS形成激发态HS*, 从而产生(公式1和2), 进而转为H2O2(公式3)。与此同时, Fe(Ⅲ)-HS络合物受光照激发发生LMCT生成Fe(Ⅱ)(公式4), 增加了浮游植物对铁的吸收, 但Fe(Ⅱ)也会被氧化为Fe(Ⅲ)(公式5),因此, HS在该光化学反应中起到催化剂的作用。其中,是活性氧形态, 既可以作为Fe(Ⅱ)的氧化剂(公式6), 也是Fe(Ⅲ)的还原剂(公式7)。
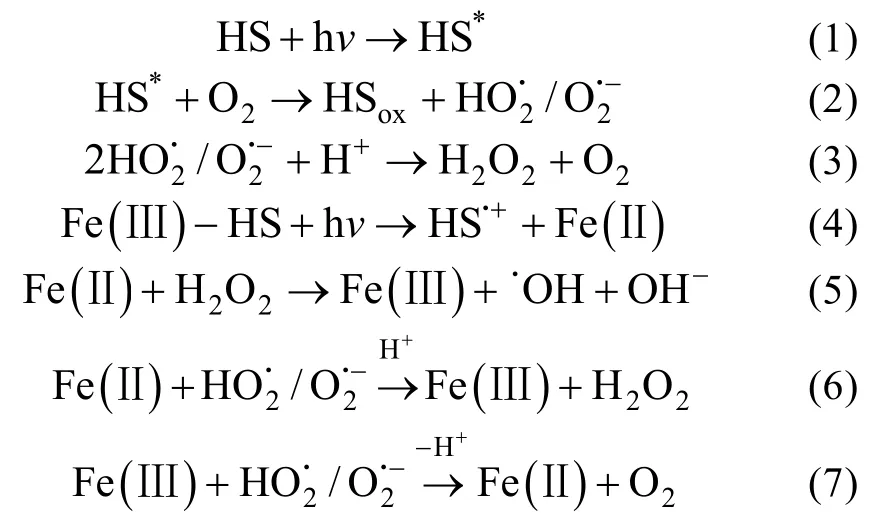
很多实验试图研究HS中发生光化学反应的结构, Miles等[74,76]认为水体中HS与Fe的氧化还原反应循环是由羧酸基团导致的。Richard等[77]发现大多数的发光基团都是HS中的低分子量部分。Ou等[78]的研究表明HS中的羧基和羟基是阿特拉津光降解的重要氧化剂。
2.4 Fe-HS络合物在河口和沿岸水域的行为
作为Fe有机配体的重要组成部分, 陆源HS在河口和沿岸区域大量存在, 因此Fe-HS络合物也大量存在。与HS和DFe类似, Fe-HS的络合强度也受到盐度的影响, 盐度越大, 络合强度越小, HS的铁配合容量(iron binding capacity, IBC)也随之降低。Fang等[43]的研究表明不同离子强度(0~0.234 g/L NaCl)下腐殖酸(HA)结构的变化对Fe-HA络合程度有较大影响, 离子强度可以影响HS的电势和空间结构, 进而影响其与Fe的络合。在高盐度条件下,Fe-HS络合物很难生成, 因为离子强度的增大降低了邻近官能团之间的静电斥力, 使HS折叠形成更致密的形状。在这种紧凑的空间结构下, HS会阻止Fe扩散到结合位点, 从而减少Fe-HS络合物的生成。离子强度对HS络合铁强度的影响表明两者之间的结合在很大程度上取决于静电作用。
Yang等[4,15]测定了长江口HA和FA在5~40盐度范围内IBC的变化, 结果表明IBC随盐度的增加而呈指数型降低, 并且符合指数衰减模型:Y(IBC,S)=A0× exp (K·S) +Y0, 其中Y(IBC,S)是HS在盐度S下的IBC值,K为IBC随盐度增加而衰减的系数,A0为长江口最大盐度下的IBC的总减少量,S为水体的盐度, 取值范围为0~34,Y0为最大盐度下的最小IBC值。Su等[4]也认为盐度影响了HS的IBC, 尤其是在长江冲淡水(YRDW)中, 盐度低的水团使得IBC升高。高盐度极大地破坏了Fe和HS的络合, 因此预测Fe和HA的络合主要发生在河流和湖泊等淡水水体中。
2.5 影响海水中HS-DFe配合的其他理化因素
Fe-HS络合物控制Fe的分布, 对海水中Fe的迁移和转化起着关键的作用[13]。除盐度外, 其他环境因素如pH值、温度等对Fe和HS之间的络合也有一定的影响, 进而影响海水中Fe-HS络合物的浓度[43,79]。
pH值也是影响Fe和HS络合的重要因素之一,随着pH的增大, HS的络合强度增大, 该现象可以用金属离子与质子对腐殖酸络合的竞争来解释[80]。在较低的pH条件下, Fe离子的水解程度较低, 丰富的游离Fe离子为HS配体提供了更多的靶离子。然而HS中酸性官能团的去质子化程度较低[80-81], 导致质子浓度较高, 与Fe争夺结合位点。此外, 在低程度去质子化的情况下, Fe容易诱导HS产生絮凝[82]。随着pH值的增加, 水解作用使溶液中的游离态铁逐渐减少, HS的去质子化成为络合的关键因素。此时HS的去质子化程度增加, 能够提供更多的结合位点并与Fe发生络合。Gledhill等[83]测定了pH值范围为6.8~8.3时河口水体中铁形态的变化, 结果表明随着pH值的降低, HS与Fe的络合会减少。除了铁离子之外, 在其他金属如钙离子[84]和铜离子[83,85]等的络合中也观察到了类似的pH效应。
温度也会影响络合强度, 过高或过低都不利于络合物的生成: 一方面, 在较低的温度下, 分子的热运动较慢, 导致络合反应速率较慢, 因此Fe-HS络合物的生成相对较少。另一方面, 由于络合物在较高温度下不稳定, 因此在较高的温度范围Fe-HS络合物的生成量减少。Fang等[43]的研究表明Fe-HA色谱峰的强度随温度的升高而增强, 在25℃时达到最大值。
此外, 金属离子之间的竞争也是影响结合位点的一个可能因素。在河口, 淡水与海水的混合增加了水的离子强度。海水中Ca和Mg的浓度之和明显高于淡水, 虽然Ca、Mg与HS的络合作用远弱于HS-Fe, 但海水中高浓度的Ca、Mg与Fe竞争HS,这可能也会影响Fe与HS的络合, 导致IBC的降低,进而影响Fe的地球化学行为。Abualhaija等[42]关于河口的研究表明HS对Fe的络合作用受到Cu竞争的影响。
3 结论及展望
腐殖质在全球溶解铁的分布中起着重要作用,是影响海水中铁的生物地球化学循环的关键性因素。目前关于海水中腐殖质与溶解态铁的研究已经取得了一定的成果, 但有些方面仍存在欠缺。比如铁结合位点数量的确定, 腐殖质具有分子多样性, 因此其铁结合位点也是可变的, 一般来说高分子量分子通常具有更多的结合位点。此外, 测量技术也存在不足, 常用电化学测定依赖于检测窗口和使用的人工配体, 所使用的人工配体不同可能会对测定结果产生影响, 如高估与腐殖质相结合的铁浓度等。为了更好的理解海水中溶解铁分布的控制因素, 除了对测量技术进行改进外, 需要在以下几个方面开展深入研究:
1) 增强对腐殖质产生和降解的认识及影响其铁结合能力过程的认识。不同来源洋流和水团的季节性变化以及浮游植物生长和消亡对腐殖质的浓度都有一定的影响, 近岸海域如长江口受到工农业废水和人类活动的影响较大, 也使得海水中的腐殖质浓度受到多种因素的影响, 研究这些影响因素是非常有必要的。
2) 收集开放海洋的数据, 研究海洋中陆源腐殖质的来源和贡献及影响其存留时间的主导控制因素。海洋中铁的生物可利用性在调节生物泵方面发挥了重要的作用, 因此腐殖质对铁的络合作用可能会影响二氧化碳的释放, 而陆源腐殖质是影响开放海洋铁含量的关键因素。因此想要了解和预测气候变化, 更好地了解陆源腐殖质的迁移及影响其铁结合能力的过程可能是重要的。
测定分析全球海域中腐殖质的浓度和分布对于理解腐殖质在海水中的存在状态、了解各种水文因素对于腐殖质的影响、进一步探索研究腐殖质对海水中铁的溶解、迁移和转化的影响有重要作用与意义。
