基于机器学习构建皮肤恶性黑色素瘤转移特征预测模型的研究
2022-06-22王英伦何孜灏黎思颖郑子豪董博文邓展程陈秋锐沈晗
王英伦,何孜灏,黎思颖,郑子豪,董博文,邓展程,陈秋锐,沈晗,2
(1.广东药科大学生命科学与生物制药学院,广东 广州 510006;2.广东省生物技术候选药物研究重点实验室,广东 广州 510006)
黑色素瘤是来源于黑色素细胞的一类高度恶性肿瘤,多发于皮肤,早期发现并手术切除原发性皮肤黑色素瘤具有较好疗效,然而其发病具有隐匿性强、易转移等特点,多数患者在晚期发生转移后才被诊断,预后较差[1]。同时,原发黑色素瘤与转移黑色素瘤在治疗手段上也存在一定差异,因此,及时准确地判断黑色素瘤的转移特征对临床治疗具有重要的指导意义。近年来,随着二代高通量测序技术的发展和应用,已积累了大量肿瘤相关的测序数据,其中以TCGA(The Cancer Genome Atlas)数据库和GEO(Gene Expression Ommibus)数据库为代表,这些数据信息为肿瘤相关研究提供了极大便利。然而,面对海量的生物信息数据,传统研究方法已无法适应要求,AI(artificial intelligence,人工智能)技术的发展让科学家可以借用电脑实现数据的高效快速分析,机器学习是AI 中最具智能特征、最前沿的研究领域之一[2]。本文选取TCGA 数据库及GEO数据库中皮肤黑色素瘤的转录组数据及临床特征信息,运用机器学习XGBoost(eXtreme Gradient Boosting)算法,构建皮肤恶性黑色素瘤转移特征预测模型,旨在为帮助判断皮肤黑色素瘤患者的临床分型及分期提供有益的借鉴。
1 方法
1.1 数据下载与整理
从TCGA 数据库网站(https://portal.gdc.cancer.gov/)下载470 例皮肤恶性黑色素瘤样本数据,包含原始RNA-Seq Count 数据、RNA 表达FPKM(Fragments Per Kilobase of exon model per Million mapped fragments)数据和临床信息数据,其中103 例为原发性肿瘤(TP)性状,367 例为转移性肿瘤(TM)性状,将FPKM 数据整理转换成TPM(Transcripts Per Kilobase of exon model per Million mapped reads)数据。从GEO 数据库网站(https://www.ncbi.nlm.nih.gov/gds/)下 载GSE190113 数 据集,其包含103 例皮肤恶性黑色素瘤样本的RNA 表达TPM 数据和临床信息数据,所有数据分析前进行标准化处理。
1.2 差异表达基因分析
分别利用R 语言(version 4.0.5)中的edgeR 包[3]与DESeq2 包[4],以TCGA 原始RNA-Seq Count 数据制成的基因表达矩阵和对应的临床信息数据作为输入资料,进行两次差异分析,完毕后,将两者所得结果以P<0.05 且|log2FoldChange|>1 为筛选阈值,获取表达差异显著基因。并将两种算法获得的差异表达基因取交集。为验证结果,使用交集差异表达基因的TPM 数据,使用R 语言中的FactoMineR包[5]进行了主成分分析(principal component analysis,PCA)。
1.3 加权基因共表达网络分析(WGCNA分析)
WGCNA 分析旨在找出协同表达的基因模块,探索基因网络与转移特征表型之间的关联,从而筛选出相关程度高的基因模块以及网络中的核心基因。利用R 语言中的WGCNA 包[6],为加快运算速度同时减少噪音,我们计算了基因表达量在样本内的方差,选取方差排名前25%的基因制成表达矩阵进行层析聚类,剔除明显离群样本。导入筛选过后的基因表达矩阵与转移相关性状(TM 与TP),选取恰当的β值作为软阈值,构建拓扑重叠矩阵,实现无标度网络。随后进行动态树剪切,将相似模块融合,完成WGCNA分析。
1.4 生物学功能和通路富集分析
利用R 语言中的clusterProfiler 包[7],对转移特征相关模块中的基因进行GO 富集分析(生物学进程BP、细胞成分CC、分子功能MF),并利用GSEA软件[8](version 4.1.0)对全部基因进行GSEA 富集分析验证GO分析结果。
1.5 ROC分析
利用R 语言中的pROC 包[9],以TP 和TM 为特征,对全部基因进行批量化ROC 分析,绘制每个基因的ROC 曲线并计算该曲线下的面积AUC 值,并筛选AUC>0.7的基因。
1.6 机器学习XGBoost分类模型的构建
对差异分析中的差异表达基因、WGCNA 分析中转移特征相关模块内的基因和ROC 分析中AUC值大于0.8 的基因进行交集分析,筛选交集作为训练模型中使用的特征。利用R语言中的mlr包[10],选择XGBoost(eXtreme Gradient Boosting,极限梯度boosting)算法并选用上述特征,构建出一个可区分转移性或非转移性恶性黑色素瘤的机器学习模型,具体方法如下:将470 个样本随机抽样分为两部分数据集,4/5 用作训练集,1/5 用作内部测试集(所有的分割数据集步骤中均确保两种类型的样本的抽取比例与原始比例相等);输入训练集,使用XG‐Boost 算法进行五折交叉验证、随机搜索1000 次的超参数调优获取最佳超参数组合,以MMCE(mean misclassification error,平均误分类误差)作为评估指标;将最优超参数组合应用于XGBoost 算法,并用其来学习训练集数据,获得机器学习分类模型;为验证包括超参数调优步骤在内的整个模型构建过程,评估该算法在训练集上的性能,采用嵌套交叉验证,其外循环为五折交叉验证,内循环为超参数调优过程。至此,获得以恶性黑色素瘤中基因表达量为特征判别非转移性或转移性的模型。最后,使用该模型预测内部测试集中以测试其泛化能力,并进一步选取GEO 数据库中的GSE190113 数据集作为外部测试集,验证该模型预测能力。
2 结果
2.1 TP组与TM组差异表达基因筛选
将TCGA 数据库来源的皮肤恶性黑色素瘤样本按转移特征分为原发肿瘤组(TP 组)和转移肿瘤组(TM 组),使用R 语言的edgeR 包与DESeq2 包进行差异分析,分别获得2041 个与3846 个差异表达基因,将两者结果取交集后获得1804 个差异表达基因。如图1所示为edgeR包分析火山图,如图2所示为DESeq2 包分析火山图,以TP 组作为对照组,蓝色点表示下调基因,红色点表示上调基因,灰色点则表示表达差异不显著的基因。交集后差异表达基因的PCA 分析结果显示TP 组和TM 组存在差异表达现象(图2)。

图1 edgeR包分析差异基因火山图Figure1 Differential gene volcano plots analyzed with the edgeR package

图2 DESeq2 R包分析差异基因火山图Figure 2 Differential gene volcano plots analyzed with the DESeq2 R package
2.2 WGCNA分析
WGCNA 分析结果显示,发现35 个转移性状相关模块(图4),其中black 模块与TM 性状的负相关性最高,r=-0.46,同时black 模块与TP 性状的正相关性最高,r=0.46,该模块包含529个基因,在TM 组中呈下调表达。

图3 交集差异表达基因的PCA分析Figure3 PCA analysis of intersection differentially expressed genes

图4 模块与转移性状的WGCNA分析Figure4 WGCNA analysis of modular and metastatic traits
2.3 GO与GSEA富集分析
GO 富集分析结果提示(图5),在生物学进程(BP)方面,black 模块中基因主要参与了表皮发育、分化等进程,在细胞成分(CC)方面,模块基因与细胞-细胞连接、角质化包膜、中间丝细胞骨架等组分密切相关,在分子功能(MF)方面,模块基因主要参与了细胞内肽酶、丝氨酸水解酶等酶活性调节及细胞骨架构建等功能。

图5 black模块基因的GO富集分析Figure 5 GO enrichment analysis of black module genes
为进一步分析转移特征基因集的分布情况,对TM 组和TP 组进行了GSEA 富集分析,图6 结果显示了在FDRq-val<0.25 条件下,标准化富集得分(NES)最高的10 个功能基因集和NES 最低的10 个功能基因集,图7 显示了与TM 组负相关的代表性GSEA 富集详细信息图,主要涉及细胞骨架中间丝的构成、黏附以及细胞连接通道活性等生物学过程,提示细胞骨架结构的减弱与肿瘤转移特征的发生有密切关系。

图6 与TM组相关的GSEA富集分析Figure 6 GSEA enrichment analysis associated with the TM group

图7 代表性的标准化GSEA富集评分(NES)与FDR值Figure 7 Representative normalized GSEA enrichment score(NES)vs.FDR values
2.4 ROC分析
基于单个基因的RNA 表达TPM 值,设定TP 与TM为判别特征,进行批量化ROC分析,绘制针对每个基因的ROC 曲线,计算其下面积AUC 值,筛选AUC>0.7的基因,共获得806个基因,利用此类基因RNA 表达TPM 值对判断TP 及TM 特征的准确性在70%以上,图8展示了代表性基因的ROC曲线。

图8 代表性基因的ROC曲线Figure 8 ROC curves of representative genes
2.5 机器学习XGBoost分类模型的建立及性能评估
基于差异表达基因、black模块基因和AUC>7.0基因,三者取交集后共获得143 个基因(图9),其中AL049555.1基因可能是污染基因,将其排除,剩余142 个基因作为机器学习的特征基因,具体基因名如下:

图9 三部分分析结果的交集基因维恩图Figure 9 Venn diagram of intersection genes of three-part analysis results



使用XGBoost 算法构建机器学习模型,为验证包括超参数调优步骤在内的整个模型构建过程,评估该算法在训练集上的性能进行的嵌套交叉验证结果显示MMCE 值约为0.12,说明该算法在训练集上表现良好。
使用构建出的分类模型对内部测试集进行预测,内部测试集共有95 例样本,预测结果绘制混淆矩阵图[10],如图10 所示,内部测试集中82 例被正确分类,13例被错误分类,MMCE值约为0.14,ROC曲线分析,AUC=0.88。
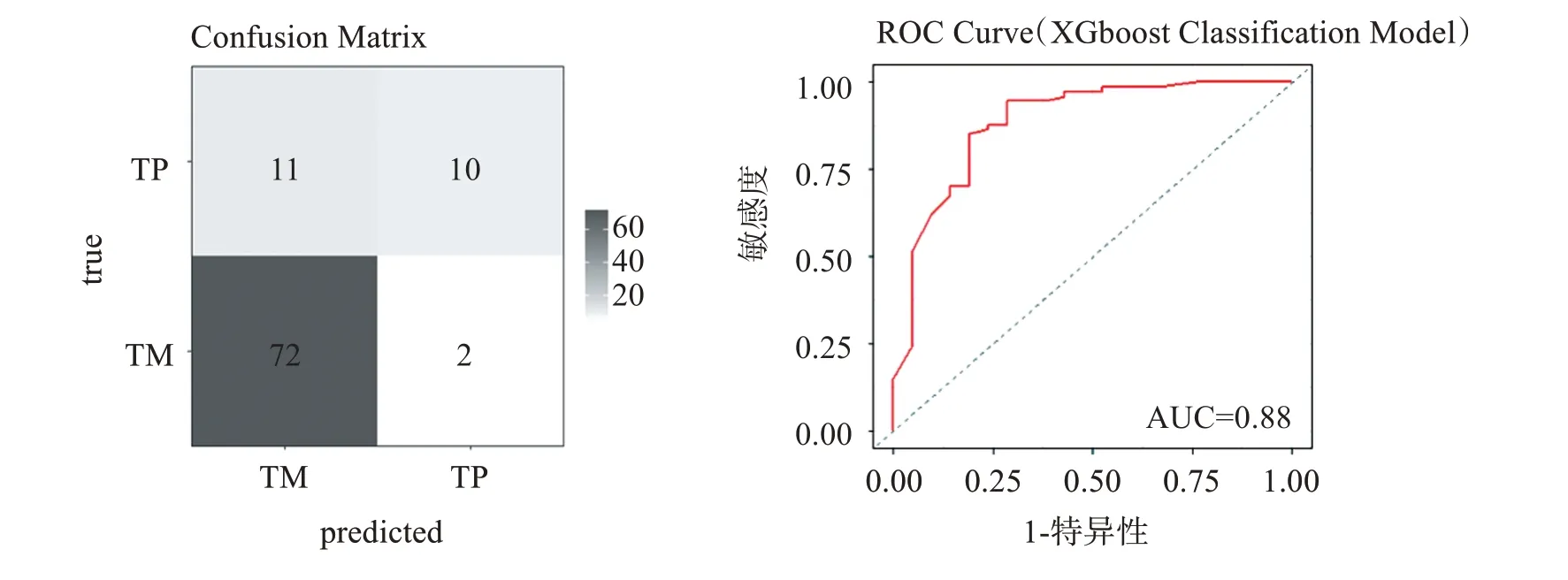
图10 内部测试集混淆矩阵及ROC曲线Figure 10 Confusion matrix and ROC curve of internal test set
使用构建出的分类模型对外部测试集GSE190113 进行预测,外部测试集共有105 例样本,如图11 所示,外部测试集中86 例被正确分类,19 例被错误分类,MMCE 值约为0.18,ROC 曲线分析,AUC=0.85。

图11 外部测试集混淆矩阵及ROC曲线Figure 11 Confusion matrix and ROC curve of external test set
3 讨论
恶性黑色素瘤占皮肤恶性肿瘤第3位,近年来,其发生率和死亡率逐年升高。由于皮肤恶性黑色素瘤易发生隐匿性转移,往往转移后病程进入中晚期,预后较差。因此,及早诊断及早局部手术切除是争取治愈的最佳方法。目前临床上,如何有效判断黑色素瘤的原发灶和转移灶特征,尚缺乏行之有效的手段。
近年来,随着生物信息技术的飞速发展,目前已累积了较为可观的肿瘤相关生物大数据,利用AI技术进行大数据分析利用,在肿瘤辅助诊断[11]、预后模型构建[12]及肿瘤标志物筛查[13]等方面已取得了长足进步。
本研究筛选TCGA 数据库中,470 例皮肤恶性黑色素瘤样本,根据其临床特征分为TP 组和TM组,应用R语言的edgeR包与DESeq2包共筛选出两组间的差异表达基因1804 个,利用WGCNA 分析筛选出与TM 及TP 性状相关性最高的black 模块,该模块包含529 个转移特征相关基因,并利用ROC曲线分析获得806 个AUC>0.7 的转移特征判别基因,3 种筛选方法取交集,最后确定了142 个转移特征基因,接着利用XGBoost 算法搭建机器学习模型,以142 个基因的RNA 表达TPM 值为输入对象,对皮肤恶性黑色素瘤的转移特征进行预测,通过内部验证集和外部验证集评估模型性能,结果证实本模型能有效判断黑色素瘤的转移特征,内部测试集的预测准确率为88%,外部验证集的预测准确率为85%。
本机器学习模型尚存在一些不足之处,首先,由于不同机构采用的检测方法存在差异,导致转录组数据可能存在批次效应,会对模型预测结果产生影响。其次,本模型采用的数据来自TCGA 数据库,样本为欧美白人人种,对于我国为代表的东亚人种是否适用尚需检验。最后,由于存在数据量和数据特征的局限性,在实际应用中可能会存在偏置性问题。因此,数据的标准化、采用国内患者样本数据及扩大数据量等措施将是本研究需要进一步改进的方向。
