有机污染物的高铁酸盐氧化去除强化技术研究进展
2022-06-22程和发
李 宇,程和发
北京大学城市与环境学院,地表过程分析与模拟教育部重点实验室,北京 100871
对于水中有机污染物的常用处理方法包括物理吸附法、化学氧化法和生物降解法,其中使用高铁酸盐〔Fe(Ⅵ)〕进行化学氧化是去除有机污染物的有效方法之一. Fe(Ⅵ)具有强氧化性,在酸性条件下的氧化还原电位(2.2 V)高于臭氧(O)、过氧化氢(HO)、氯气(Cl)、二氧化氯(ClO)和高锰酸钾(KMnO)等常用水处理氧化剂,并且在较宽的pH 范围内具有较强的氧化能力. 相比于基于无选择性的羟基自由基(OH)的高级氧化技术(AOPs),Fe(Ⅵ)的选择性高,易于氧化富含供电子基团的有机污染物,受环境基质影响较小,Fe(Ⅵ)与Cl、HCO、NO、SO等常见阴离子和Na、K等常见阳离子的反应活性均较低,低浓度的腐殖酸(HA)还能对Fe(Ⅵ)氧化有机污染物起到促进作用,且Fe(Ⅵ)能够在实际污水处理的pH (pH 为6.0~9.0)下进行反应. 虽然Fe(Ⅵ)能够有效氧化分子结构中含有不饱和官能团(如酚羟基、芳胺基和碳碳双键)的有机污染物,但其在酸性条件下易于自分解,且选择性高,因此Fe(Ⅵ)对许多不饱和度较低的有机污染物反应活性低. 此外,在碱性条件下,Fe(Ⅵ)氧化能力也会降低. 因此,Fe(Ⅵ)直接氧化法亟需改进,以扩大其适用范围、提高其氧化能力,使其适用于实际污水处理.
近年来,国内外学者开发了诸多Fe(Ⅵ)氧化的强化技术,以提高Fe(Ⅵ)对有机污染物的处理效能,去除Fe(Ⅵ)本身难以氧化的有机污染物,并取得了较大进展. 根据Fe(Ⅵ)氧化的强化体系所产生的活性物种的类别,可将其分为基于高价铁氧中间体〔Fe(Ⅳ)/Fe(Ⅴ)〕的Fe(Ⅵ)活化技术和基于活性自由基(如OH 和SO)等其他活性物种的Fe(Ⅵ)协同技术. Fe(Ⅵ)的活化及协同技术可以提高Fe(Ⅵ)与有机污染物的反应速率、Fe(Ⅵ)的利用效率和有机污染物的降解率,有效去除与Fe(Ⅵ)反应活性低的有机污染物,同时减少Fe(Ⅵ)用量. 但由于在Fe(Ⅵ)活化或协同体系中起作用的活性物种发生变化,进而改变有机污染物的降解动力学和降解机理,以及降解产物的种类、分布和毒性,因此需要全面深入地对Fe(Ⅵ)的活化及协同技术进行评估和研究. 在已有研究基础上,该文对近年来开发的Fe(Ⅵ)活化及协同技术的研究进展进行系统性总结,梳理Fe(Ⅵ)直接氧化有机污染物的处理效能、反应动力学和氧化机理、Fe(Ⅵ)的活化技术及协同技术氧化有机污染物的效能与机理,讨论Fe(Ⅵ)活化及协同技术引起的有机污染物降解产物种类、分布及毒性变化,并对未来研究方向进行展望,以期为Fe(Ⅵ)在实际水处理中的高效利用提供参考方法.
1 Fe(Ⅵ)直接氧化有机污染物
1.1 处理效能
Fe(Ⅵ)直接氧化法已经广泛用于去除水中有机污染物,并展现出良好的处理效能. Sun 等在pH=8.0 时使用30 μmol/L 的Fe(Ⅵ)氧化降解2 μmol/L的苯胺,发现150 s 时即可达到92%的去除率;杨滨等在pH=7.0 时使用Fe(Ⅵ)氧化降解三氯生,发现Fe(Ⅵ)与三氯生的浓度比大于7∶1 时,2 μmol/L 的三氯生即可得到完全去除;黄军磊等在pH=7.0 时使用300 μmol/L 的Fe(Ⅵ)氧化降解20 μmol/L 的吲哚美辛,发现20 min 时可达到95%的去除率;Feng等在pH=7.0 时使用Fe(Ⅵ)氧化降解30 μmol/L 的氟喹诺酮类抗生素,发现在Fe(Ⅵ)与目标污染物的浓度比为20∶1 时,在2 min 内即可完全去除恩诺沙星、诺氟沙星、氧氟沙星和马波沙星4 种抗生素. 综上,Fe(Ⅵ)可在数分钟内基本完全去除含有供电子基团的有机污染物,具有良好的处理效能.
1.2 反应动力学
Fe(Ⅵ)和大部分有机污染物(以X 表示)的氧化还原反应都遵循二级反应动力学模型,即
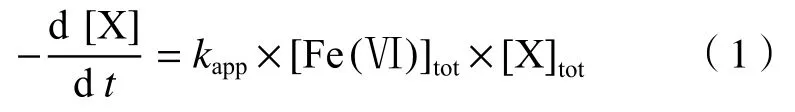
式中:为Fe(Ⅵ)与有机污染物反应的表观二级动力学反应速率常数,L/(mol·s);[Fe(Ⅵ)]为不同离子形态Fe(Ⅵ)的总浓度,mol/L;[X]为不同离子形态有机污染物的总浓度,mol/L. Fe(Ⅵ)的解离常数(p)为1.6、3.5 和7.2,主要以HFeO、HFeO、HFeO和FeO四种形态存在.通常是在Fe(Ⅵ)浓度远低于有机污染物浓度的条件下,使Fe(Ⅵ)浓度变化符合一级动力学特征,使用停流光谱技术测定Fe(Ⅵ)浓度的变化速率,进而计算得到的,一般在10~10L/(mol·s)范围内.
Fe(Ⅵ)氧化取代基不同的苯环取代化合物时,与取代基之间存在结构-活性关系. 例如,Sun等研究表明,Fe(Ⅵ)和取代苯胺化合物反应的的对数值与取代基系数呈线性关系;Fe(Ⅵ)氧化苯并三唑类化合物和多氯联苯硫化物的的对数值均与取代基系数存在线性自由能关系. 通过Fe(Ⅵ)和苯环取代化合物反应的的对数值与取代基系数之间的线性自由能关系,可以使用已知Fe(Ⅵ)与苯环取代化合物的和取代基系数等参数,预测Fe(Ⅵ)与其他结构类似但取代基不同的苯环取代化合物反应的.
1.3 氧化机理
Fe(Ⅵ)对有机污染物的直接氧化遵循单电子(1 e)或双电子(2 e)转移机制,因此其对有机污染物的氧化具有高选择性,电子转移机理如图1 所示.Fe(Ⅵ)的选择性氧化导致其反应活性受到目标有机污染物的结构与性质的影响,Fe(Ⅵ)易于和含有富电子官能团的有机物(如有机硫化合物、胺类化合物、酚类化合物等)发生反应,但对于醇类化合物、醛类化合物、糖类化合物等反应活性较低. Fe(Ⅵ)氧化有机污染物的选择性可以体现在Fe(Ⅵ)与有机污染物反应的的差异上,如Xie 等对比研究了Fe(Ⅵ)氧化两种苯胂酸类化合物对氨基苯胂酸和洛克沙胂的动力学特征,发现分子中含有供电子基团氨基的对氨基苯胂酸与Fe(Ⅵ)反应的远高于分子中含有吸电子基团硝基的洛克沙胂. Jiang 等使用Fe(Ⅵ)处理环丙沙星和布洛芬的混合溶液,发现环丙沙星的去除率明显高于布洛芬,这是由于环丙沙星分子中的供电子基团更多. Yang 等使用Fe(Ⅵ)处理污水处理厂二级出水中的68 种有机污染物,发现Fe(Ⅵ)能够选择性地氧化降解富含供电子基团的有机污染物,如具有酚类结构的雌激素和三氯生,具有双键结构的卡马西平、雄激素、孕激素和糖皮质激素,以及含仲铵基的酸性药物和含苯胺基的抗生素,而Fe(Ⅵ)与布洛芬、非诺洛芬、环磷酰胺等供电子基团较少的有机污染物的反应活性较低.
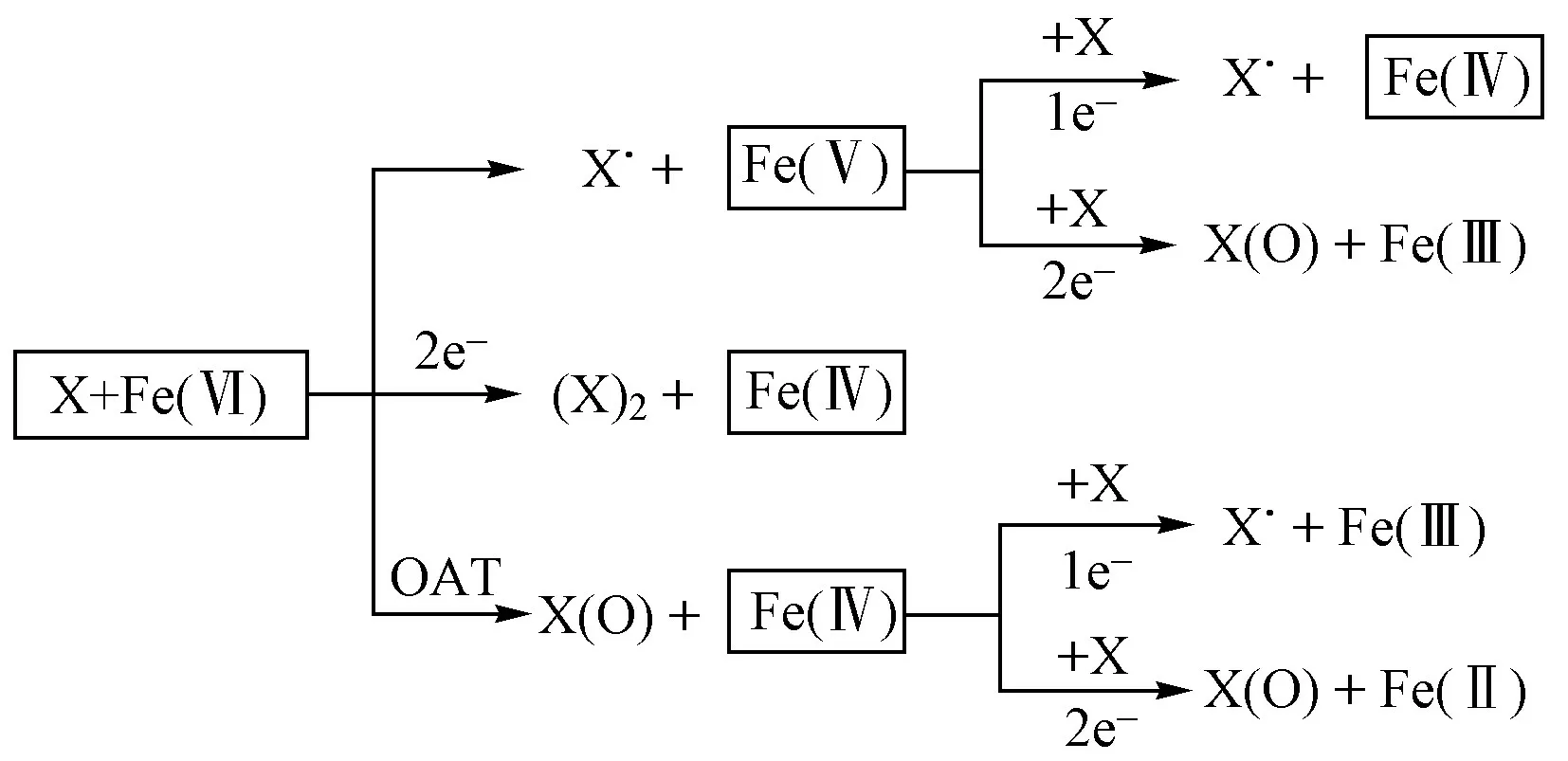
图 1 Fe(Ⅵ)直接氧化有机污染物(以X 表示)的电子转移机理示意[16]Fig.1 Diagram of the electron transfer mechanism during the oxidation of organic pollutant (represented by X) by Fe(Ⅵ)[16]
与有机污染物反应时,Fe(Ⅵ)首先被还原为中间价态的Fe(Ⅳ)/Fe(Ⅴ),Fe(Ⅳ)/Fe(Ⅴ)性质不稳定,会快速自分解或进一步通过电子转移与有机污染物反应.Fe(Ⅵ)与有机污染物的反应机理通常包括:①先通过单电子转移从Fe(Ⅵ)转化为Fe(Ⅴ),之后Fe(Ⅴ)进一步氧化有机污染物;②通过双电子转移转化为Fe(Ⅳ)和二聚体,之后Fe(Ⅳ)进一步氧化有机污染物;③通过氧原子转移(OAT)生成Fe(Ⅳ)和加氧产物,之后Fe(Ⅳ)进一步氧化有机污染物. 此外,Fe(Ⅵ)、Fe(Ⅴ)、Fe(Ⅳ)也会发生自分解反应,最终生成Fe(Ⅱ)和Fe(Ⅲ). 有机污染物被Fe(Ⅵ)氧化的降解路径取决于其具体化学结构,不同的化学结构和官能团导致其降解机理和降解路径不同,需要通过O 示踪法和产物分析法对Fe(Ⅵ)氧化有机污染物的机理进行深入研究.
Fe(Ⅵ)直接氧化法对含有不饱和官能团的有机污染物的氧化能力较强,但存在一定局限性,如在酸性条件下Fe(Ⅵ)会迅速自分解,而在碱性条件下Fe(Ⅵ)的高稳定性导致有机污染物降解缓慢. 此外,Fe(Ⅵ)氧化的高选择性导致其与不饱和官能团较少的有机污染物的反应活性较低. 因此,Fe(Ⅵ)直接氧化法亟需进行改进与强化,从而开发出一系列Fe(Ⅵ)的活化及协同技术.
2 Fe(Ⅵ)活化技术
研究表明,中间价态的Fe(Ⅳ)/Fe(Ⅴ)与有机化合物反应的比Fe(Ⅵ)高2~6 个数量级,因此可以使用化学方法将Fe(Ⅵ)转化为Fe(Ⅳ)/Fe(Ⅴ)以促进有机污染物的降解. Fe(Ⅵ)的活化技术通过在中性或偏碱性条件下,向Fe(Ⅵ)氧化体系中投加活化剂,诱导低活性的Fe(Ⅵ)生成高活性的Fe(Ⅳ)/Fe(Ⅴ),从而提高有机污染物的降解速率和降解率. 根据投加的活化剂在反应体系中的状态,可将Fe(Ⅵ)活化技术分为均相活化技术和异相活化技术.
2.1 均相活化技术
Fe(Ⅵ)的均相活化技术包括酸活化Fe(Ⅵ)、还原剂活化Fe(Ⅵ)等.
2.1.1 酸活化Fe(Ⅵ)
Manoli 等研究发现,向偏碱性的Fe(Ⅵ)和有机污染物混合体系中加入少量酸时,pH 变化很小,但咖啡因、安赛蜜、阿替洛尔的降解率提高了30%左右,并且所需反应时间大幅降低,他们推测酸活化Fe(Ⅵ)降解有机污染物的活性物种为Fe(Ⅳ)/Fe(Ⅴ).Manoli 等还发现,Fe(Ⅵ)氧化降解咖啡因受到水中Ca和Mg的抑制作用,而酸活化Fe(Ⅵ)可以减轻Ca和Mg对咖啡因降解的抑制作用,虽然酸活化前后咖啡因的降解都受到天然有机物(NOM)和二级污水的抑制作用,但Fe(Ⅵ)直接氧化咖啡因并不矿化,而酸活化Fe(Ⅵ)在NOM 存在和二级污水反应体系中均有明显矿化. Manoli 等使用酸活化Fe(Ⅵ)处理实际污水处理厂出水中的多种有机污染物,发现相比于Fe(Ⅵ)直接氧化,污水中药物的去除率提高了12.6%~56.2%,因此酸活化Fe(Ⅵ)技术更适用于实际水处理. 然而,以上研究均未直接观测到酸活化Fe(Ⅵ)后产生的活性物种,因此酸活化Fe(Ⅵ)生成Fe(Ⅳ)/Fe(Ⅴ)的假设还需进一步试验验证.
2.1.2 还原剂活化Fe(Ⅵ)
由于过渡金属元素Fe 具有多种价态(0、+2、+3、+4、+5 和+6),因此可以通过还原剂的还原作用将Fe(Ⅵ)转化为强氧化性的Fe(Ⅳ)/Fe(Ⅴ),促进有机污染物的降解. Feng 等研究了多种无机还原剂对Fe(Ⅵ)的活化作用,发现还原剂可以在30 s 内有效活化Fe(Ⅵ)降解有机污染物,且降解效能与还原剂的种类和用量密切相关. Fe(Ⅵ)直接氧化甲氧苄氨嘧啶的降解率为16%,双电子转移还原剂〔如NHOH、As(Ⅲ)、Se(Ⅳ)、P(Ⅲ)和NO〕可 将Fe(Ⅵ)还 原 为Fe(Ⅳ),将降解率提高至20%~40%,而单电子转移还原剂(如SO)则将Fe(Ⅵ)还原为Fe(Ⅴ),将降解率提高到100%. Fe(Ⅱ)、Mn(Ⅱ)、Fe(Ⅲ)等还原剂也可以有效活化Fe(Ⅵ),实现对有机污染物的高效去除. Dong 等研究发现,2,2-联氮-二(3-乙基-苯并噻唑-6-磺酸)二铵盐(ABTS)可以在较宽的pH 范围(6.0~10.0)内活化Fe(Ⅵ)生成Fe(Ⅴ)和氧化性的ABTS,促进双氯芬酸的降解. pH=8.0 时,Fe(Ⅵ)直接氧化体系和Fe(Ⅵ)/ABTS 体系降解双氯芬酸的准一级降解速率分别为0.085 和3.08 min. 在pH 为7.0~9.0 范围内,HO也可以作为还原剂,通过双电子转移活化Fe(Ⅵ)生成Fe(Ⅳ)〔Fe(Ⅵ)+HO→Fe(Ⅳ)+O,=21.18 L/(mol·s) (pH=8.0)〕,并通过链式反应生成Fe(Ⅴ),促进多种有机污染物的降解,其机理如图2所示. pH=8.0 时,Fe(Ⅵ)直接氧化体系和Fe(Ⅵ)/HO体系降解磺胺甲恶唑的准一级降解速率分别为0.08和0.30 min.
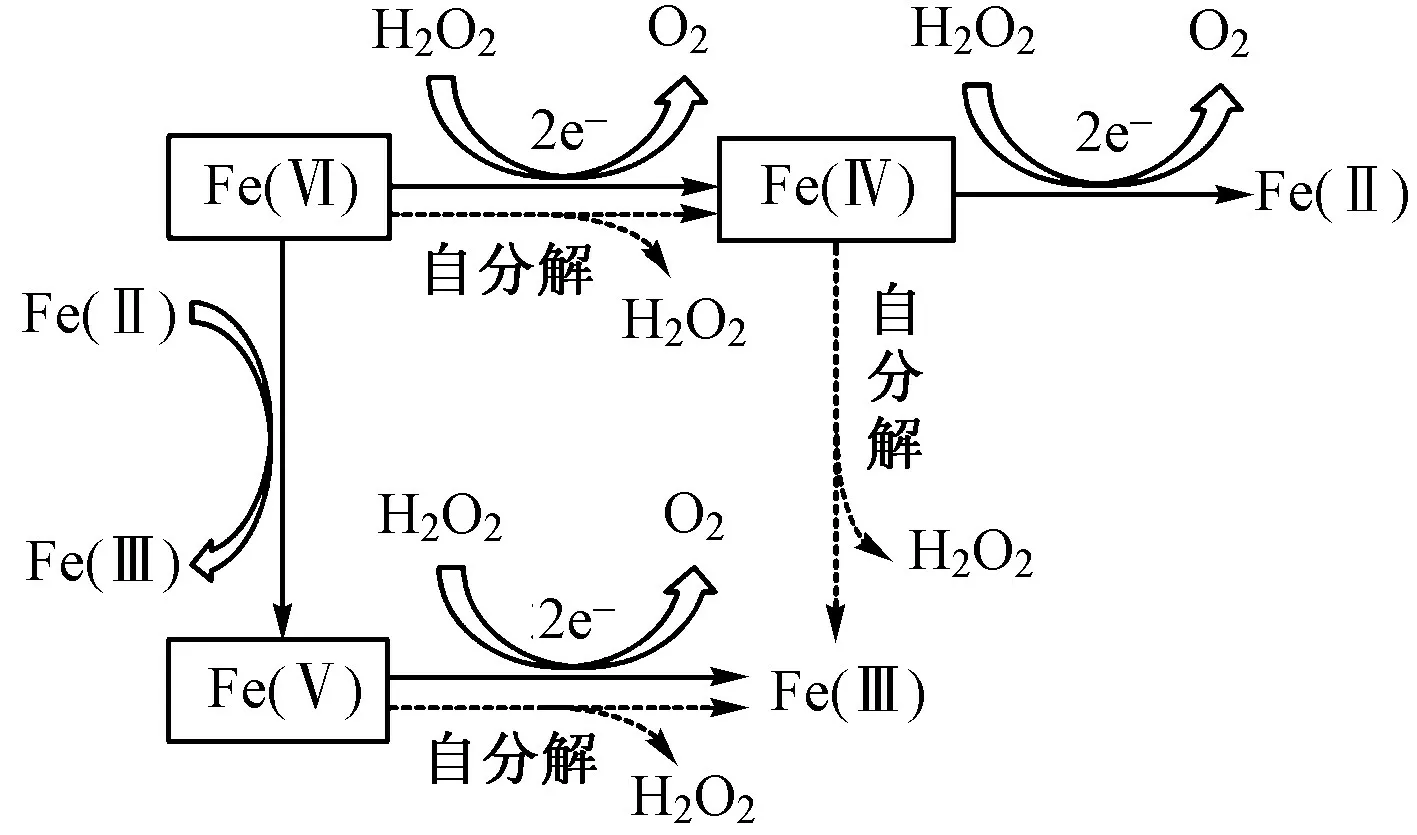
图 2 H2O2 活化Fe(Ⅵ)机理示意[32]Fig.2 Diagram of the mechanisms of Fe(Ⅵ) activation by H2O2[32]
相比于Fe(Ⅵ)直接氧化,还原剂活化Fe(Ⅵ)大多只能在反应初始阶段促进有机污染物的氧化降解,且还原剂自身也会消耗产生的Fe(Ⅳ)/Fe(Ⅴ),因此Fe(Ⅵ)的整体氧化容量可能反而降低. 与其他还原剂不同,氨氮可以提高Fe(Ⅵ)与有机污染物的反应速率和有机污染物降解的表观速率,而不只是在初始阶段提高有机污染物的降解率. Feng 等研究发现,向Fe(Ⅵ)氧化体系加入氨氮后,Fe(Ⅵ)氧化氟甲喹的速率可以提高5~12 倍,具体数值取决于氨氮的浓度和pH. 这可能是由于氨氮可以通过与Fe(Ⅵ)的中间产物Fe(Ⅳ)/Fe(Ⅴ)络合,提高Fe(Ⅳ)/Fe(Ⅴ)的反应活性,从而提高氟甲喹的准一级降解速率.
2.2 异相活化技术
异相活化技术是指向Fe(Ⅵ)与目标有机污染物的混合体系中投加不溶于水的固体颗粒,通过一系列固体表面作用或缓释活化剂实现活化Fe(Ⅵ)的技术,已开发的异相活化技术包括二氧化硅(SiO)活化Fe(Ⅵ)、过氧化钙(CaO)活化Fe(Ⅵ)、碳质材料活化Fe(Ⅵ)等.
2.2.1 SiO活化Fe(Ⅵ)
Manoli 等研究发现,在pH 约为8.0 时,使用4 g/L 的SiO可将Fe(Ⅵ)直接氧化咖啡因的降解率由53.0%升至100%,且增加SiO用量可以减轻NOM对咖啡因降解的抑制作用. 活化机理可能为SiO抑制Fe(Ⅱ)/Fe(Ⅲ)和Fe(Ⅵ)/Fe(Ⅴ)的反应,从而增加Fe(Ⅵ)的暴露量,使更多的Fe(Ⅵ)能与有机污染物反应. 此外,Fe(Ⅵ)可能会吸附在SiO凝胶表面,Fe 与Si 的作用能够产生强氧化剂〔如Fe(Ⅳ)〕,并且降低Fe(Ⅵ)的自分解,从而促进咖啡因的氧化.
2.2.2 CaO活化Fe(Ⅵ)
Zhang 等使用CaO缓释HO,以活化Fe(Ⅵ)降解磺胺甲恶唑,发现磺胺甲恶唑在Fe(Ⅵ)直接氧化体系和Fe(Ⅵ)/CaO体系中的降解率分别为35.4%和82.7%. 促进效果可能是由于CaO缓释HO,一方面将Fe(Ⅵ)活化为Fe(Ⅳ),并通过链式反应生成Fe(Ⅴ);另一方面抑制了HO与Fe(Ⅳ)/Fe(Ⅴ)的副反应〔Fe(Ⅳ)+HO→Fe(Ⅱ)+O,=10L/(mol·s) (pH=7.0);Fe(Ⅴ)+HO→Fe(Ⅲ)+O,=5.07×10L/(mol·s) (pH=8.0)〕,从而提高了Fe(Ⅵ)和HO的利用率,促进有机污染物的降解. 此外,Fe(Ⅳ)/Fe(Ⅴ)在偏碱性条件下的自分解符合二级动力学特征,即Fe(Ⅳ)/Fe(Ⅴ)的浓度越高,其自分解越剧烈,因此CaO可以通过缓释HO活化Fe(Ⅵ),降低Fe(Ⅳ)/Fe(Ⅴ)的浓度,从而抑制其自分解,提高有机污染物的降解率.
2.2.3 碳质材料活化Fe(Ⅵ)
碳质材料是一类能够用于去除水体中有机污染物的催化材料,表面具有丰富的活性位点,可以活化多种氧化剂. Sun 等研究发现,碳纳米管(CNT)可以在较宽pH 范围(7.0~10.0)内加速Fe(Ⅵ)降解溴苯酚类化合物,在pH=8.0 时,加入25、50、100 mg/L 的CNT 可将3-溴苯酚降解的准一级速率常数由Fe(Ⅵ)直接氧化体系的0.003 4 s分别升至0.005 9、0.011、0.016 s,推测活化机理为CNT 表面酚羟基与Fe(Ⅵ)反应生成高活性的Fe(Ⅳ)/Fe(Ⅴ). Fe(Ⅵ)/CNT体系能够选择性氧化富电子有机污染物(如溴苯酚类化合物、磺胺甲恶唑等),但对少电子的有机污染物(如碘帕醇、硝基苯等)的反应活性低,体现了Fe(Ⅳ)/Fe(Ⅴ)的氧化选择性. Pan 等进一步比较研究了多种碳质材料对Fe(Ⅵ)的活化能力,发现碳质材料本身不吸附或少量吸附有机污染物,但可以通过表面的羰基活化Fe(Ⅵ)生成Fe(Ⅳ)/Fe(Ⅴ),促进有机污染物的降解. 碳质材料的活化能力与其种类有关,水热炭的活化能力最强. 但由于碳质材料本身也可能存在环境风险和毒性危害,因此后续仍需对碳质材料活化Fe(Ⅵ)技术进行安全风险评估. 生物炭是环境友好的碳质材料,是由生物质在厌氧条件下热解形成的,具有成本低、表面含氧官能团丰富等优点,并且具有良好的催化性能. Tian 等研究发现,生物炭也可以活化Fe(Ⅵ)有效降解有机污染物,加入生物炭后,Fe(Ⅵ)对5 种有机污染物(磺胺甲恶唑、卡马西平、环丙沙星、双氯芬酸、N,N-二乙基-3-甲基苯甲酰胺)的氧化速率提高了3~14 倍,TOC 的去除率增加了2.4~8.0 倍,因此生物炭活化Fe(Ⅵ)技术是水处理和污水处理中有潜力的氧化方法. 通过甲基苯基亚砜探针化合物的试验表明,Fe(Ⅳ)/Fe(Ⅴ)是起作用的活性物种,推测机理为生物炭表面的供电子基团(如酚羟基)将电子转移给Fe(Ⅵ),活化Fe(Ⅵ)生成高活性的Fe(Ⅳ)/Fe(Ⅴ),从而促进有机污染物的降解.
综上,Fe(Ⅵ)的异相活化技术主要是通过固体活化剂的表面作用或缓释活化剂将Fe(Ⅵ)活化为高活性的Fe(Ⅳ)/Fe(Ⅴ). 因此,可以寻找具有特定官能团或具有缓释能力的固体活化剂,开发新型异相活化技术. Fe(Ⅵ)的活化技术具有受到基质影响小、反应速率快等优点,其机理如图3 所示. 但对于某些Fe(Ⅵ)难以氧化的有机污染物,活化后可能也难以去除. 而Fe(Ⅵ)协同技术可以通过产生高活性的自由基(如OH、SO)等活性物种,有效去除与Fe(Ⅵ)反应活性低的有机污染物.

图 3 Fe(Ⅵ)活化技术机理示意Fig.3 Diagram of the mechanisms of Fe(Ⅵ)activation technologies
3 Fe(Ⅵ)协同技术
Fe(Ⅵ)的协同技术通过将Fe(Ⅵ)氧化法和紫外光或协同剂相结合,实现对有机污染物的协同去除.相比于Fe(Ⅵ)活化技术,Fe(Ⅵ)协同技术可能会产生Fe(Ⅳ)/Fe(Ⅴ)之外的新的活性物种,如多种活性自由基(OH、SO、O)等. 与Fe(Ⅵ)的活化技术不同,Fe(Ⅵ)协同技术不局限于中性及偏碱性条件,在酸性条件下也可以实现Fe(Ⅵ)的协同作用. Fe(Ⅵ)的协同技术主要包括Fe(Ⅵ)/氧化剂协同、Fe(Ⅵ)/含硫还原剂协同、Fe(Ⅵ)/紫外光协同和Fe(Ⅵ)/光催化协同.
3.1 Fe(Ⅵ)/氧化剂协同
Fe(Ⅵ)可与多种常用水处理氧化剂〔如过硫酸氢盐(PMS)、过硫酸盐(PDS)等〕产生协同作用,促进有机污染物的降解,协同作用的机理为Fe(Ⅵ)的还原产物与氧化剂反应生成活性自由基 (如OH、SO).Feng 等使用Fe(Ⅵ)/PMS 体系降解4 种氟喹诺酮类化合物(FQs),发现向Fe(Ⅵ)和有机污染物混合体系中投加PMS 后,相比于Fe(Ⅵ)直接氧化,氟甲喹、恩诺沙星、马波沙星、氧氟沙星的降解率分别提高了42%、15%、24%和28%. PMS 可以将Fe(Ⅵ)的还原产物Fe(Ⅲ)还原为Fe(Ⅱ),之后Fe(Ⅱ)催化PMS产生SO,促进FQs 的降解. Wu 等使用Fe(Ⅵ)/PMS体系氧化降解阿特拉津,发现Fe(Ⅵ)/PMS 体系能够在较宽pH 范围(5.0~9.0)内有效去除阿特拉津. pH=6.0 时,Fe(Ⅵ)直接氧化体系和Fe(Ⅵ)/PMS 体系中阿特拉津的降解率分别为11.7%和81.5%. 电子顺磁共振结果和自由基抑制试验表明,Fe(Ⅵ)/PMS 体系可以产生OH 和SO,而SO是导致阿特拉津降解的主要活性物种. 此外,除Fe(Ⅲ)外,Fe(Ⅵ)的还原产物氧化铁(FeO)也可以活化PMS 生成SO,促进阿特拉津的降解. Li 等发现,Fe(Ⅵ)/PDS 体系可以协同降解抗生素环丙沙星,投加羟胺可以加速Fe(Ⅲ)的还原和Fe(Ⅱ)的再生,从而进一步提高Fe(Ⅵ)/PDS体系对环丙沙星的降解率. Fe(Ⅵ)直接氧化体系、Fe(Ⅵ)/PDS 体系和Fe(Ⅵ)/PDS/羟胺体系中环丙沙星的降解率分别为54.2%、72.6%、91.5%,Fe(Ⅵ)及产生的OH 和SO是Fe(Ⅵ)/PDS 体系的主要活性物种.
3.2 Fe(Ⅵ)/含硫还原剂协同
亚硫酸盐(SO)是优良的还原剂,Feng 等研究发现,Fe(Ⅵ)/SO体系可以在15 s 内提高难降解抗生素甲氧苄啶及氟甲喹的降解率,并推测在该体系中,多种活性物种〔Fe(Ⅳ)/Fe(Ⅴ)、SO、SO和OH〕可能参与氧化反应,有机污染物的降解率取决于溶液pH 和[SO]/[Fe(Ⅵ)] (浓 度 比). Sun 等研 究发现,Fe(Ⅵ)/SO体系可以在10 s 内降解约78%的N,N-二乙基-3-甲苯酰胺,而Fe(Ⅵ)直接氧化不发生降解,并确认SO为主要活性物种. 而Zhang 等使用Fe(Ⅵ)/SO2氧化降解磺胺甲恶唑,发现OH 和SO是主要活性物种. 为了准确鉴定此体系中起作用的活性物种,Shao 等选取难溶的CaSO作为SO的缓释源,研究了Fe(Ⅵ)-CaSO体系的氧化效能并对活性物种进行了鉴定,发现Fe(Ⅵ)-CaSO体系的氧化机理主要是溶解的SO2通过单电子转移直接将Fe(Ⅵ)还原为Fe(Ⅳ)/Fe(Ⅴ). 该体系可以有效氧化多种有机污染物(磺胺甲恶唑、恩诺沙星、卡马西平、双氯芬酸钠、阿特拉津、布洛芬和苯甲酸),氧化速率比Fe(Ⅵ)直接氧化快6.1~173.7 倍,且对实际水体中的有机污染物也有较好的去除效果. 综上,Fe(Ⅵ)/SO2体系虽然具有快速去除有机污染物的潜力,但不同研究中主要活性物种不同,这可能是由试验条件不同所致,如SO2与Fe(Ⅵ)浓度比的变化可使活性物种发生变化. Shao 等研究发现,在Fe(Ⅵ)/SO2体系中,活性物种的生成情况与SO2与Fe(Ⅵ)的浓度比相关. 当SO2与Fe(Ⅵ)的浓度比为0.1~0.3 时,Fe(Ⅴ)为主要活性物种;当SO2与Fe(Ⅵ)的浓度比为0.4~1.0 时,Fe(Ⅴ)、OH 和SO为主要活性物种;当SO2与Fe(Ⅵ)的浓度比为1.5~10.0 时,OH 和SO为主要活性物种. 此外,相比于单次投加,分次投加SO2可以进一步提高有机污染物的去除效能.
除SO2外,硫代硫酸盐(SO)也可以协同Fe(Ⅵ)促进有机污染物降解,但其活性物种存在争议. Zhang等将Fe(Ⅵ)与SO2结合,发现该体系能够协同降解氯霉素,主要氧化活性物种为OH 和SO的作用,而Gao 等研究结果表明Fe(Ⅵ)/SO2体系起作用的活性物种只有Fe(Ⅳ)/Fe(Ⅴ). 因此,对于Fe(Ⅵ)/SO体系的活性物种仍需进一步探究.
3.3 Fe(Ⅵ)/紫外光协同
紫外照射(UV)是水处理中常用的手段,Aslani等发现Fe(Ⅵ)/UV 体系能够协同去除卤乙酸,推测Fe(Ⅵ)、Fe(Ⅴ)、OH 为主要活性物种. 而Wu 等使用Fe(Ⅵ)/UV 体系降解2,4-二氯苯酚,发现Fe(Ⅵ)/UV体系降解2,4-二氯苯酚的速率常数分别比UV 体系和Fe(Ⅵ)氧化体系高6.9 和9.2 倍,O为主要活性物种. 张雄军等使用UV 和Fe(Ⅵ)结合降解双酚A,发现Fe(Ⅵ)/UV 体系具有明显协同作用,与Fe(Ⅵ)直接氧化相比,协同体系的COD去除率提高了15%左右(UV 单独作用时去除率为1.5%). 该体系的协同作用是由于UV 提高了双酚A 的反应活性,使其更易于被Fe(Ⅵ)降解,并且Fe(Ⅵ)在自分解过程中产生的HO和UV 构成了UV/HO高级氧化体系,从而促进双酚A 的降解. 综上,Fe(Ⅵ)/UV 体系的机理存在争议,仍需进一步探究.
3.4 Fe(Ⅵ)/光催化协同
Fe(Ⅵ)/光催化体系研究主要集中于Fe(Ⅵ)/UV/TiO体系. Fe(Ⅵ)和光催化的协同氧化是利用TiO光催化(UV/TiO)产生的导带电子还原Fe(Ⅵ),得到高活性的Fe(Ⅴ). 由于Fe(Ⅵ)接收了导带电子,减少了导带电子和空穴的复合,从而提高空穴的氧化效率. 空穴协同Fe(Ⅴ)、Fe(Ⅵ)氧化水中的有机污染物,从而实现协同效果. Yuan 等将Fe(Ⅵ)与UV/TiO光催化体系相结合,发现Fe(Ⅵ)/UV/TiO体系可以协同降解邻苯二甲酸二甲酯. pH=9.0 时,邻苯二甲酸二甲酯在Fe(Ⅵ)直接氧化体系中几乎不降解,在UV/TiO体系中的降解率为12%,而在Fe(Ⅵ)/UV/TiO体系中的降解率达到了65%,体现了明显的协同作用.
综上,Fe(Ⅵ)的活化技术是通过生成高活性的Fe(Ⅳ)/Fe(Ⅴ)对Fe(Ⅵ)直接氧化进行强化,而Fe(Ⅵ)的协同技术则是通过生成活性自由基(如OH、SO、O等)对Fe(Ⅵ)直接氧化进行强化,协同技术的机理如图4 所示. Fe(Ⅵ)活化及协同技术可以提高Fe(Ⅵ)对有机污染物的降解率及降解速率,降低Fe(Ⅵ)用量,去除与Fe(Ⅵ)反应活性低的有机污染物,因此Fe(Ⅵ)活化及协同技术既可以保留Fe(Ⅵ)氧化不易受到环境基质影响、产物具有吸附絮凝能力的优点,还能够通过生成高活性的氧化物种提高Fe(Ⅵ)的氧化能力与氧化容量,加快有机污染物的降解并提高其降解率. Fe(Ⅵ)的活化技术可以降低环境基质对Fe(Ⅵ)的影响,不足之处是Fe(Ⅵ)活化产生的Fe(Ⅳ)/Fe(Ⅴ)可能也难以氧化与Fe(Ⅵ)反应活性低的有机污染物,因为Fe(Ⅳ)/Fe(Ⅴ)对有机污染物的氧化机理与Fe(Ⅵ)类似,都是通过电子转移进行的.Fe(Ⅵ)协同技术可以生成高活性的自由基(如OH、SO等),氧化Fe(Ⅵ)不能直接降解的有机污染物,不足之处是活性自由基的低选择性会导致其受到环境基质的影响较大. 因此,需要根据目标有机污染物种类和水体基质成分选择合适的Fe(Ⅵ)活化或协同技术进行处理. Fe(Ⅵ)的活化及协同技术对有机污染物降解的促进效果与活性物种见表1.

图 4 Fe(Ⅵ)协同技术机理示意Fig.4 Diagram of the mechanisms of Fe(Ⅵ)synergy technologies
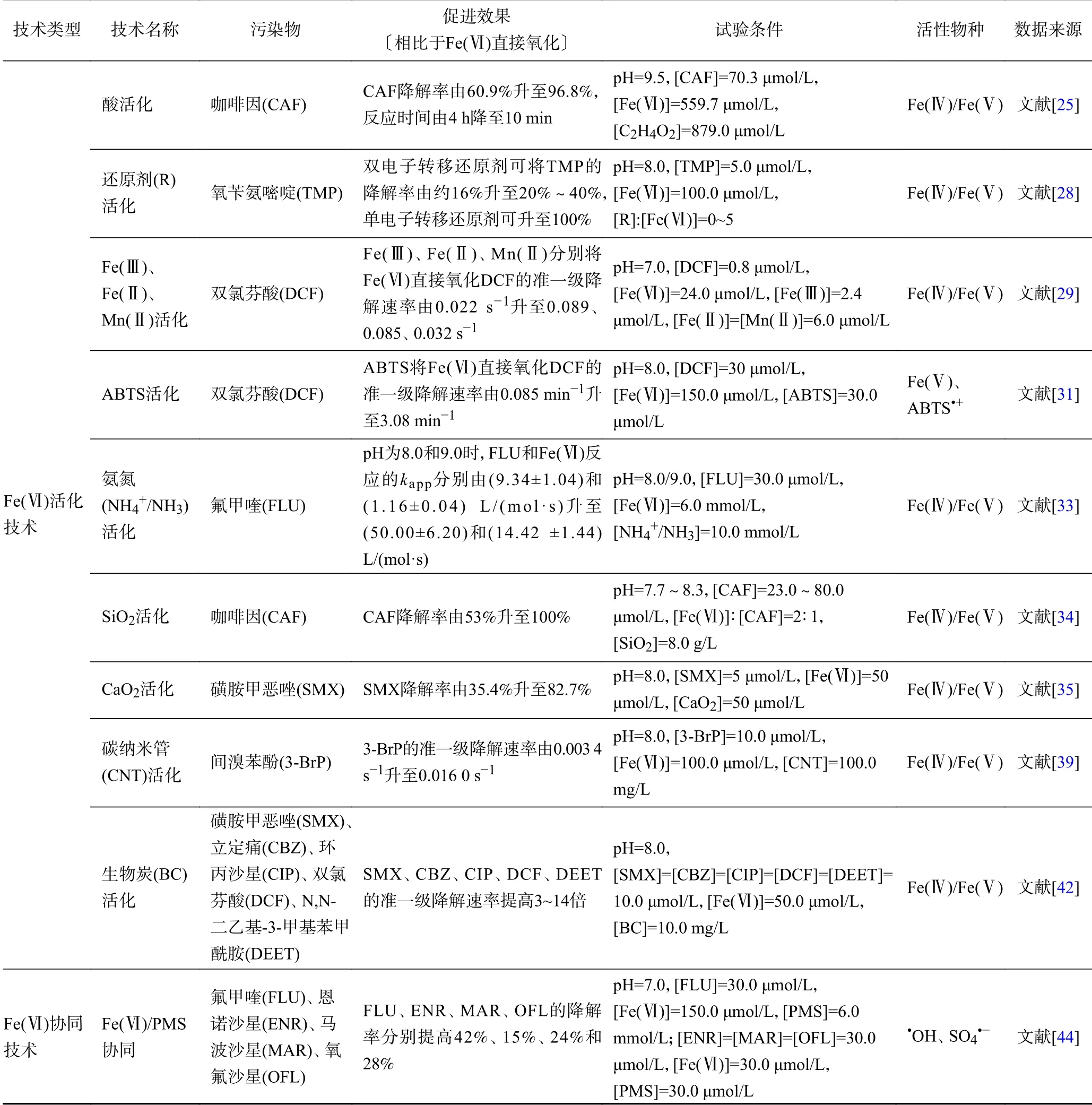
表 1 Fe(Ⅵ)活化及协同技术对有机污染物降解的促进效果与活性物种Table 1 The promotion effect on the degradation of organic pollutants and active species by Fe(Ⅵ) activation and synergy technologies
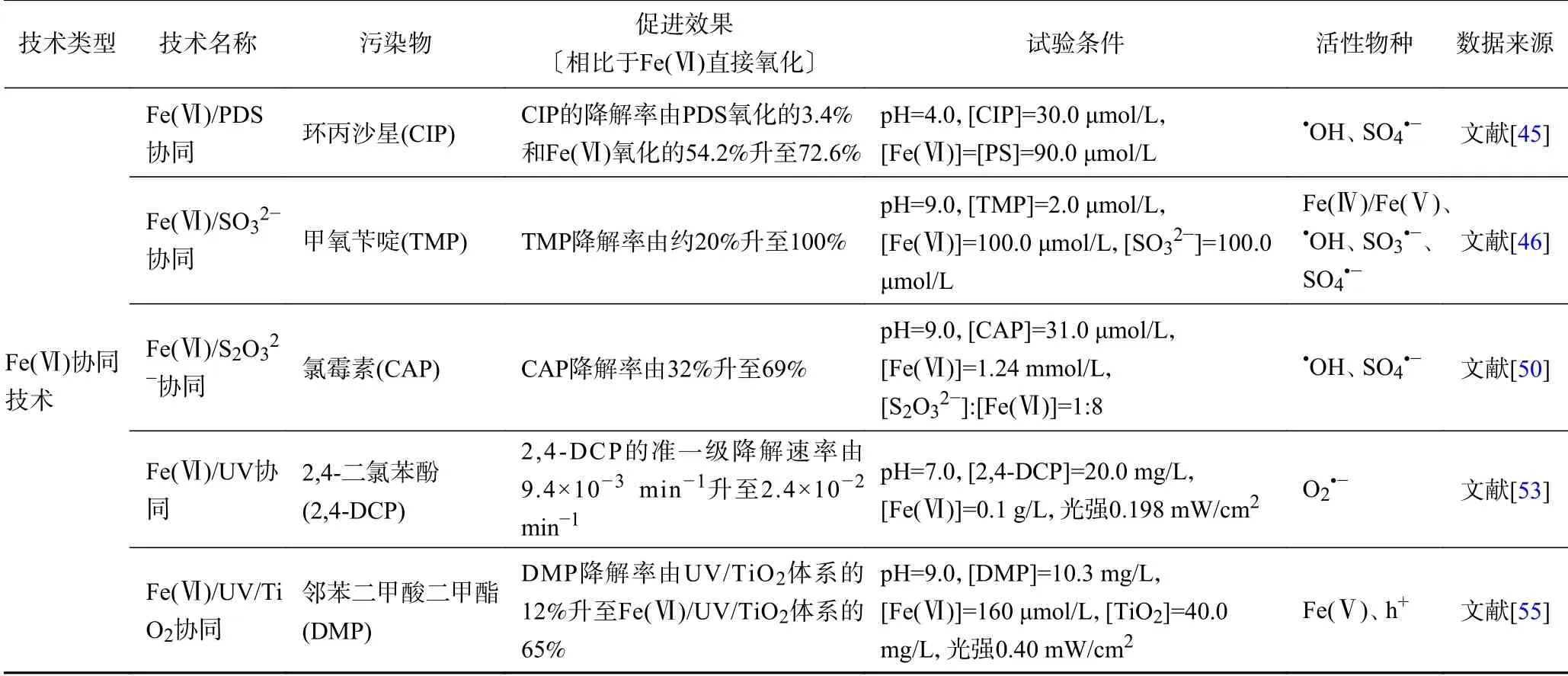
续表 1
4 Fe(Ⅵ)氧化的强化技术对有机污染物降解产物种类、分布及毒性影响
同种有机污染物经过不同技术处理,其降解产物种类、分布及毒性都可能发生变化. 由于Fe(Ⅵ)活化及协同技术是通过产生新的活性物种对有机污染物进行降解,因此降解机理、降解产物的种类、分布和毒性等都可能有所变化. 现有的Fe(Ⅵ)活化及协同技术更多关注于有机污染物的降解效能及Fe(Ⅵ)活化或协同技术的机理探究,对有机污染物的降解产物及其毒性变化研究较少.
相比于Fe(Ⅵ)直接氧化法,Fe(Ⅵ)的活化技术可生成高活性的Fe(Ⅳ)/Fe(Ⅴ). Fe(Ⅳ)/Fe(Ⅴ)与Fe(Ⅵ)氧化有机污染物的机理均为电子转移,但其自身性质可能不同于Fe(Ⅵ),从而改变有机污染物的降解产物. Feng 等使用氨氮活化Fe(Ⅵ)氧化降解氟甲喹时发现,与Fe(Ⅵ)直接氧化相比,氟甲喹降解产物的种类不变,但产物分布发生变化. 这是因为氨氮络合的Fe(Ⅳ)/Fe(Ⅴ)更易于进攻氟甲喹的C=C键,从而导致氨氮存在时,碳碳双键断裂的产物在液相色谱-质谱联用仪中测定的峰面积明显升高,而相应的羟基化产物的峰面积明显下降. Fe(Ⅵ)协同技术是通过产生新的活性物种(如活性自由基等)促进有机污染物的降解,Fe(Ⅵ)与协同试剂的浓度比及投加方式均可能对有机污染物的降解产物带来影响. Shao 等发现,使用Fe(Ⅵ)/SO2协同体系降解恩诺沙星的产物与SO与Fe(Ⅵ)的浓度比有关,在不同浓度比下,有不同产物生成. Gong 等将紫外光与Fe(Ⅵ)/PMS协同体系相结合降解磺胺甲恶唑,发现Fe(Ⅵ)、OH和SO是主要的活性物种,且不同试剂投加方式对磺胺甲恶唑降解产物的种类和分布有所影响. 单次投加Fe(Ⅵ)时,磺胺甲恶唑的主要降解路径为硝基化、羧基化、脱甲基、羟基化和苯环的开环;分步投加Fe(Ⅵ)时,磺胺甲恶唑的主要降解路径为羟基化、羧基化和S—N 键的断裂;而分步投加PMS 时,磺胺甲恶唑的主要降解路径为羟基化和羧基化. 这是由于不同试剂投加方式可能导致活性物种的比例发生变化,从而改变磺胺甲恶唑降解产物的种类及分布. 相比于Fe(Ⅵ)直接氧化法,Fe(Ⅵ)协同体系生成的自由基(如OH、SO)氧化能力更强,且选择性低,因此可能生成更多Fe(Ⅵ)直接氧化不能生成的降解产物. 因此,Fe(Ⅵ)协同体系除Fe(Ⅵ)直接氧化的降解途径以外,还会有新的降解途径,降解产物种类总体是增加的. 例如,Feng 等使用Fe(Ⅵ)/PMS 协同体系降解氟甲喹,发现相比于Fe(Ⅵ)直接氧化,协同体系检测到了3 种脱氟产物,可能是SO作用导致的降解路径.
目前对于Fe(Ⅵ)活化前后反应体系毒性对比的相关研究十分有限. Cao 等使用Fe-苯酚改性生物炭活化Fe(Ⅵ)降解阿特拉津,通过发光细菌急性毒性测试评估反应前后溶液毒性的变化,反应30 min后,相比于阿特拉津原始溶液,Fe(Ⅵ)直接氧化体系的抑制率降低了11.38%,Fe-苯酚改性生物炭活化Fe(Ⅵ)体系的抑制率降低了19.02%,表明Fe-苯酚改性生物炭活化Fe(Ⅵ)体系对阿特拉津溶液毒性降低效果更明显. Zhang 等使用CaO缓释HO,以活化Fe(Ⅵ)降解磺胺甲恶唑,发现反应结束后,磺胺甲恶唑的原始溶液、Fe(Ⅵ)氧化体系、CaO活化Fe(Ⅵ)体系的抑制率分别为81.66%、68.78%和71.36%,表明CaO活化Fe(Ⅵ)体系与未活化体系反应后的毒性接近,两种体系均能有效降低磺胺甲恶唑溶液的毒性.
Fe(Ⅵ)活化及协同技术可以导致活性物种发生变化,从而影响有机污染物的降解产物种类和分布,进而影响反应体系的毒性. 研究Fe(Ⅵ)活化及协同技术氧化有机污染物的产物及毒性变化情况对评价该技术是否适用于实际水体处理十分重要,但目前有关Fe(Ⅵ)活化及协同技术降解有机污染物的毒性变化研究十分有限,需进一步加强相关研究.
5 结论与展望
a) 在Fe(Ⅵ)活化体系中,Fe(Ⅵ)转化为高活性的Fe(Ⅳ)/Fe(Ⅴ),实现对有机污染物的快速氧化. 但一方面,均相活化体系的活化剂浓度越高,产生Fe(Ⅳ)/Fe(Ⅴ)的速率越快,Fe(Ⅳ)/Fe(Ⅴ)的自分解也越剧烈;另一方面,使用还原剂活化Fe(Ⅵ)时,过量的还原剂会竞争消耗产生的Fe(Ⅳ)/Fe(Ⅴ),从而降低活化效果,如HO活化Fe(Ⅵ)时,过量的HO会竞争消耗Fe(Ⅳ)/Fe(Ⅴ),而使用CaO缓释HO可以提高Fe(Ⅵ)的活化效果. 因此通过具有缓释能力的异相活化剂,如开发固体酸催化剂缓释H以活化Fe(Ⅵ),可以减缓Fe(Ⅳ)/Fe(Ⅴ)的生成,抑制其自分解,减少还原剂对Fe(Ⅳ)/Fe(Ⅴ)的消耗,进而提高Fe(Ⅵ)活化技术去除有机污染物的效能.
b) Fe(Ⅵ)活化及协同技术能够明显提高Fe(Ⅵ)氧化有机污染物的效能,缩短反应时间. 但相关研究主要关注于对有机污染物降解的促进效果,部分技术〔如酸活化Fe(Ⅵ)技术〕的机理未知或存在争议,需对Fe(Ⅵ)的活化及协同机理进行进一步探究.
c) Fe(Ⅵ)活化及协同技术可以生成新的活性物种,从而对有机污染物降解产物的种类、分布及毒性产生影响. 目前对有机污染物降解产物变化的相关研究较少,需深入探究Fe(Ⅵ)活化及协同技术对有机污染物降解产物种类、分布及毒性的影响.
d) Fe(Ⅵ)活化及协同技术的相关研究主要在纯水体系中进行,对于实际废水中的有机污染物去除研究较少. 实际废水基质复杂,有机污染物种类多、浓度低,未来应加强对实际废水中有机污染物的处理效能研究.
