CRISPR/Cas技术在乳酸菌中的研究进展
2022-06-15房思昌宋馨薛玉玲王世杰
房思昌,宋馨,薛玉玲,王世杰,*
1(河北科技大学 食品与生物学院,河北 石家庄,050018)2(上海理工大学 医疗器械与食品学院,上海,200093) 3(石家庄君乐宝乳业有限公司,河北 石家庄,050221)
1987年,日本研究者在对大肠杆菌iap基因测序时发现了一段异常结构,这一结构串联间隔重复。重复基因由29个碱基构成,非重复基因由32个碱基构成,在其他原核生物中没有发现与其同源的基因序列。由于当时受到科学技术的制约及人们认识的匮乏,没有深入了解其生物学功能[1]。随后,有学者研究发现间隔重复序列广泛存在于原核生物基因组和古细菌中,将它们命名为成簇的规则间隔短回文序列(clustered regularly interspaced short palindromic repeats,CRISPR),并且发现CRISPR是可移动的序列,在含有CRISPR的原核生物中还发现了比较保守的基因称为Cas基因[2]。此外,在化脓链球菌(StreptococcuspyogenesSF370)中发现CRISPR序列的间隔区与其他菌株中的前噬菌体具有高度的同源性,且噬菌体不能入侵具有相同CRISPR序列间隔区的古细菌细胞,由此推测CRISPR序列可能对外源DNA具有特异性免疫功能。
随着人们对原核生物基因组分析需求的增加,研究者进一步将基因组学应用到细菌中并阐明其生物学功能[3]。2007年,BARRANGOU团队在对嗜热链球菌基因组分析时首次证实了CRISPR系统能够特异性抵御噬菌体的入侵[4],2010年以后,CRISPR-Cas系统的功能逐渐明确:由向导RNA引导Cas蛋白酶靶向切割外源DNA的特异性免疫系统。CRAWLEY等[5]在对1 262种乳酸菌基因组分析时发现普遍存在CRISPR免疫功能,其中Ⅱ-A型系统在自身乳酸菌宿主中占主要活性,能够有效对外源及宿主DNA进行编辑,SANOZKY-DAWES等[6]在对加氏乳杆菌(Lactobacillusgasseri)CRISPR/Cas研究时发现存在一种Ⅱ-A型系统,其间隔区呈现多样性,证实了该系统能够应用在乳酸菌中,拉开了应用CRISPR进行乳酸菌功能解析的序幕。
1 CRISPR-Cas系统
CRISPR-Cas系统由一个CRISPR基因序列和相对保守的Cas基因组成,存在于大约83%的古细菌和45%的细菌中。CRISPR基因座由前导序列(leader),重复序列(repeat)和间隔序列(spacer)构成。前导序列位于CRISPR基因座上游,长度为500~700 bp,是CRISPR转录的起始点,重复序列长度为28~37 bp,负责转录反式激活CRISPR RNA(tracrRNA)形成发卡结构。间隔序列是从侵入的外源DNA中截取的一小段基因序列,相当于将入侵者拉入了“黑名单”,当携带有此间隔序列的外源DNA再次侵入时,CRSPR-Cas系统会进行精确的识别并清除[7]。Cas基因负责转录Cas蛋白,Cas蛋白包括Cas1~Cas10等多种蛋白,负责间隔序列的获取和对外源DNA的防御,其中CRISPR/Cas9系统发展速度最快且在乳酸菌中应用最为广泛[5]。
CRISPR-Cas9系统中Cas9蛋白、成熟的CRISPR RNA(crRNA)和tracrRNA、以及原间隔区相邻基序(protospacer adjacent motif,PAM)是最主要的4种元件,Cas9蛋白包括2个易于识别的核酸酶结构域HNH和RUVC,就像两把剪刀将DNA的两条单链进行剪切;crRNA是间隔序列的转录产物,它由RNA内切酶Ⅲ(RNaseⅢ)促进成熟并与tracrRNA一起形成单向导RNA(single guide RNA,sgRNA),其作用为引导Cas9蛋白到正确的外源DNA切割位点[8];当然,只靠sgRNA的引导作用是不够的,还需要另一关键因素PAM序列作为特异性的识别位点,它是位于外源DNA上由NGG(N为任意碱基)3个碱基组成的小段序列,Cas9复合物与PAM近端结合,DNA双链解开[9]。CRISPR-Cas9简单免疫流程为:Cas1、Cas2等蛋白对入侵的外源DNA进行剪切并将其一段基因序列放入CRISPR序列中,再次入侵时CRISPR序列识别并转录产生Cas9蛋白、crRNA、tracrRNA,2种RNA结合形成sgRNA与Cas9蛋白形成效应复合物并识别PAM序列近端双链DNA使其解旋,Cas9核酸酶2个结构域分别切割形成双链断裂(double strand break,DSB)(图1)。随后出现了Cas9的突变体nickase Cas9(nCas9)与dead Cas9(dCas9),nCas9又名Cas9切口酶,它与Cas9蛋白的差异在于失活了HNH(Cas9D10A)或RUVC(Cas91840A)结构域,在被单向导RNA引导到靶向位置后单一的结构域能够在靶基因单链形成一个小的缺口而不是DSB,更易实现断
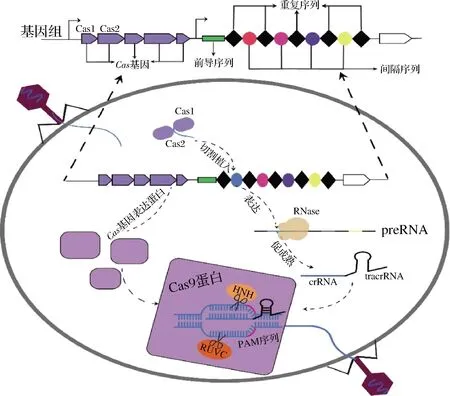
图1 CRISPR-Cas9基因座及免疫流程Fig.1 CRISPR-Cas9 locus and immune process
裂基因的修复,目前已在干酪乳杆菌(Lactobacilluscasei)中得到了很好的应用[10]。dCas9中2个核酸酶结构域都失活使其失去双链切割的能力,但依然能够与双链结合,阻碍了RNA聚合酶与靶基因的结合,成为实现基因沉默的重要工具,并在植物乳杆菌(Lactiplantibacillusplantarum)和乳酸乳球菌(Lactococcuslactis)中得到良好的应用[11-12]。
2 CRISPR-Cas系统分类
CRISPR系统按基因座和Cas蛋白可以分为两大类(表1);一类包括Ⅰ、Ⅲ、Ⅳ型,二类包括Ⅱ、Ⅴ、Ⅵ型,每种类型因作用位点不同又分为多种亚型,包含标志性蛋白来进行靶基因的切割,不同的是二类系统使用单一的多结构域蛋白发挥作用,而一类系统使用大型的多亚基复合体来进行切割,这种大亚基蛋白由CRISPR基因座上的Cas7蛋白形成螺旋骨架连接Cas5并与crRNA紧密结合,Cas8或Cas10及其他亚基与Cas5连接。一类系统通常由Cas6或Cas5负责促使crRNA成熟,二类系统中Ⅱ型系统crRNA的成熟靠RNaseⅢ,Ⅴ、Ⅵ型大部分crRNA成熟分别靠Cas12、Cas13效应蛋白的部分核酸酶活性物质催化。切割过程中Ⅰ型Cas8负责PAM的定位并由Cas3解旋酶切割,Ⅲ型系统通过效应复合物中Cas10的CARF结构域靶向入侵的DNA和RNA,Ⅳ型属于Ⅰ型与Ⅲ型的衍生物,因缺乏必要的Cas蛋白其潜在的功能还不明确。Ⅱ、Ⅴ、Ⅵ型主要通过Cas9、Cas12a、Cas13及相关结构域HNH、Ruvc、HEPN进行剪切[13-14]。在174种乳酸菌的基因组分析中,58个基因组中发现了CRISPR序列,其中Ⅱ型系统在链球菌和乳杆菌中较为丰富、在副干酪乳杆菌(Lactobacillusparacasei)、戊糖乳杆菌(Lactobacilluspentosus)中发现了Ⅰ型系统的存在,其次是Ⅲ型系统,109个基因组中未检测到CRISPR序列,其他类型如Ⅳ型、Ⅴ型和Ⅵ型系统在基因组中没有检测到[15]。
受乳酸菌物种差异的影响,可以将其分为内源性与外源性CRISPR系统,内源性系统指自身存在的完整系统,在嗜热链球菌中较为常见,但因基因组中缺乏非同源末端连接(non-homologous end joining,NHEJ)而不能得到很好的利用,HIDALGO-CANTABRANA等[21]分析了卷曲乳杆菌(Lactobacilluscrispatus)中内源性Ⅰ型系统的作用机制,并成功实现了p-gtf(胞外多糖引导糖基转移酶基因)的敲除,为重新利用内源性系统提供了依据。利用外源性的CRISPR系统是目前有效的方法,可以在一些缺乏完整的CRISPR系统如嗜酸乳杆菌(Lactobacillusacidophilus)、巴氏乳杆菌(Lactobacilluspasteurii)或含有内源性系统的乳酸菌中得到利用,GOH等[22]在利用外源性nCas9系统pLCNICK嗜酸乳杆菌进行了基因的敲除和插入,同时证实可以在加氏乳杆菌和副干酪乳杆菌中得以利用。
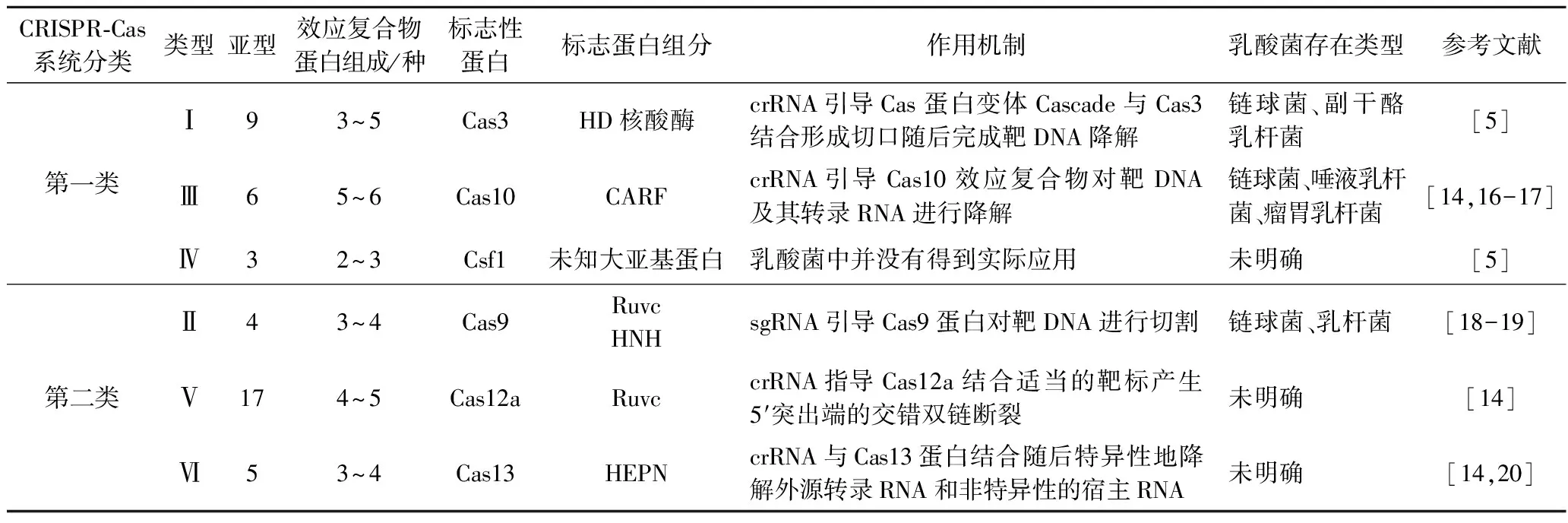
表1 CRISPR-Cas系统分类及作用机制Table 1 Classification and action mechanism of CRISPR CAS system
3 CRISPR-Cas9在乳酸菌中的应用
3.1 基因编辑工具
3.1.1 传统基因编辑工具
目前已经开发了多种乳酸菌基因编辑技术:基于质粒的同源重组交换技术、线性DNA重组技术和基于质粒的CRISPR基因编辑技术。
基于质粒的同源重组有单交换和双交换2种方法,单交换方法指质粒与目的基因两侧的同源序列一次交换实现基因的敲除与插入,LELOUP等[23]利用pRV300敲除质粒在清酒乳杆菌(Lactobacillussake)上通过同源重组单交换置换掉pstⅠ基因及lacL基因,阐释了2种基因生物学存在的意义。但这种方法重组效率低,容易引进抗性基因,增加后期筛菌的难度,双交换的方法利用二次同源交换解决了抗性基因遗留的问题,GROOT等[24]利用双交换法在植物乳杆菌WCFS1分析了与Mn2+稳态之间的关系。应用2次同源交换剔除单交换插入的目的基因,但这种编辑方法操作繁琐且耗时,成本高。
线性DNA重组技术与基于质粒的同源重组技术原理相同,包含单链DNA重组和双链DNA重组,都是利用同源序列进行基因组的同源交换,不同的是线性DNA重组技术不需要构建质粒,只需在外源重组酶及类似物的辅助线性化为单链DNA完成与目的基因的交换,因此不会引入除目标基因以外的其他基因,例如抗性基因,但此项技术在乳酸菌中并没有广泛展开,原因是乳酸菌缺乏辅助该技术的外源重组酶及类似物[25]。
3.1.2 基于CRISPR-Cas9的基因编辑技术
基于CRISPR的编辑技术已经成为乳酸菌基因组编辑的强力工具,CRISPR-Cas9的简便操作性使其成为分子生物领域中的热点。目前已经开发了基于Cas9和Cas9变体的编辑技术以及与传统基因编辑工具联用的技术。
CRISPR-Cas9辅助的线性DNA重组技术在线性重组的基础上利用了Cas9剪切致死性效果,提高了后期对野生型菌株的筛选效率。OH等[26]首次在乳酸杆菌中实现了基于CRISPR编辑的第一个示例CRISPR-Cas9选择与单链DNA(ssDNA)重组相结合并成功在罗伊氏乳杆菌(Lactobacillusreuteri)ATCC6475中进行了编辑。通过SpyCas9靶向野生型序列清除未编辑的细胞,成功地编辑了基因组中的3个不同位点,筛选出克隆的效率高达100%。GUO等[27]首次在乳酸乳球菌NZ900中利用高效外源重组酶Rect优化了ssDNA重组,并结合Cas9反选择消除未突变野生型菌株,在72 h内效率达到75%以上。ZHOU等[28]在植物乳杆菌WCSF1中利用Cas9线性DNA重组技术在无抗情况下通过核糖体替换和启动子的点突变增强了前体途径,实现了n-乙酰氨基葡糖的产生。
HUANG等[29]通过RecE/T辅助修复的Cas9工具,在双质粒系统中实现植物乳杆菌WCSF1和短乳杆菌ATCC 367单基因敲除,敲除效率为50%~100%,高效修复双链断裂。SONG等[10]在干酪乳杆菌(Lactobacilluscasei)中失活Cas9的D10A结构域形成单链缺口的Cas9D10A系统,有效绕过DSB的致死性,与前者相比,后者更有效刺激同源重组修复。SONG等[30]以pLL质粒为基础在乳酸乳球菌NZ9000中开发了单质粒CRISPR系统,经过启动子的筛选和单向导sgRNA的替换后对ldh基因进行了有效的敲除,敲除效率达50%。此外,将Cas9结构域同时失活构成的dCas9系统,其中的dCas9蛋白与目标基因结合后失去靶向裂解基因的能力,成为实现基因沉默和转录调控的强大工具。XIONG等[11]在乳酸乳球菌NZ9000中开发了基因转录抑制的双质粒系统,使用诱导型启动子Pnisin驱动链球菌中dCas9的表达,而强的组成型启动子P44驱动引导RNA表达单个或多个基因,效率高达99%,成为基因沉默的重要工具。目前已成功应用到大肠杆菌、芽孢杆菌、链球菌、葡萄球菌和乳酸乳球菌中。一系列Cas9变体的出现成为创造乳酸菌Cas9工具箱新路径。CRISPR-Cas基因编辑技术如表2所示。
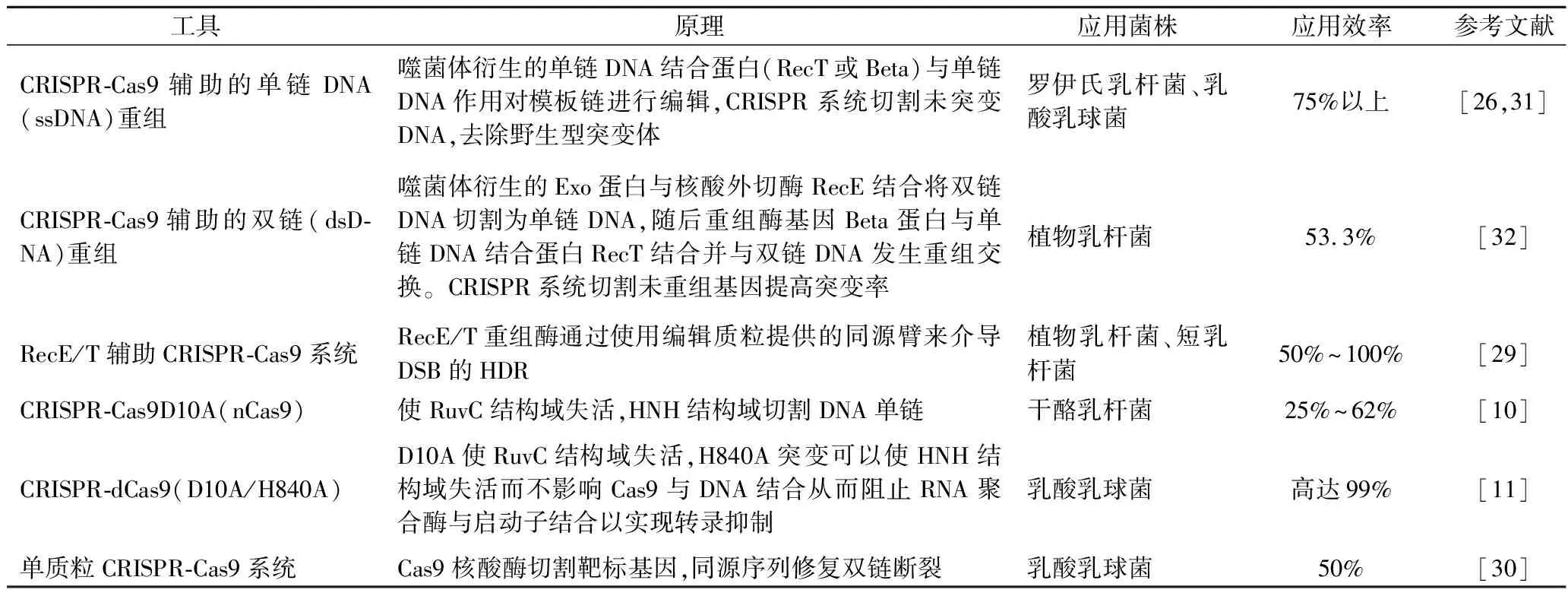
表2 CRISPR介导的操作技术Table 2 CRISPR mediated operational techniques
3.2 CRISPR-Cas9技术的应用
3.2.1 功能基因解析
基因编辑是利用一定的手段对目的基因进行删除、插入和替换的操作技术。由于Ⅱ型CRISPR-Cas9系统在乳酸菌中占据多数,最常用的来源于化脓链球菌CRISPR-Cas9(spyCas9)系统已应用到干酪乳杆菌、植物乳杆菌、罗伊式乳杆菌、嗜热链球菌、乳酸乳球菌中。LEMAY等[33]利用SpyCas9在乳酸乳球菌MG1363中研究噬菌体侵入与细菌蛋白质组成的关系,通过敲除llmg_0219基因产生了对p2噬菌体的抗性,揭示了乳酸菌抗性的原因。WANG等[34]利用Spycas9基因编辑系统构建了植物乳杆菌AR113单、双、三重bsh(胆盐水解酶基因)敲除突变体,通过对4种bsh基因的敲除,利用不同种类的胆汁酸盐评价bsh突变后的菌株耐胆盐活性,得出bsh1与bsh3对胆盐耐受性至关重要,甘氨酸结合的胆盐是bsh的首选底物,为胆汁酸耐受性高的菌株的合理高通量筛选提供了依据。MYRBRÅTEN等[12]利用SpyCas9在植物乳杆菌WCSF1中通过双质粒dCas9系统探索了细菌生长过程中Acm2、DnaA和EzrA(细胞周期基因)相关蛋白的表达含量,通过抑制3种基因的细胞分离蛋白的表达得出DnaA、EzrA与细胞的生长周期有着密切的联系。TIAN等[35]利用SpyCas9系统以副干酪乳杆菌NCBIO01-M2ldh(乳酸脱氢酶基因)为靶标对其进行敲除和过表达,筛选出在45 ℃下产量达221 g/L、光学与化学强度达99%的L-乳酸菌株(表3)。
3.2.2 CRISPR基因座利用
CRISPR间隔物反映了菌株的演变轨迹,当菌株被外源噬菌体侵入时,新的间隔物会按时间顺序整合到CRISPR序列的前导区,由于CRISPR-Cas在许多乳酸菌中占据较高的发生率,在菌株分型和CRISPR分类方面提供了一定的数量优势。PUJATO等[37]在49株乳杆菌中分析了其间隔子含量、CRISPR序列及Cas蛋白的分布,通过PCR筛选后发现29株菌中含有完整的CRISPR系统,Ⅱ型丰富度最高,仅在副干酪乳杆菌85中发现Ⅰ型CRISPR系统且该系统较为活跃。ROGALSKI等[38]利用CRISPR间隔序列长度成功在发酵面团中分离出11种旧金山乳杆菌(Lacto-
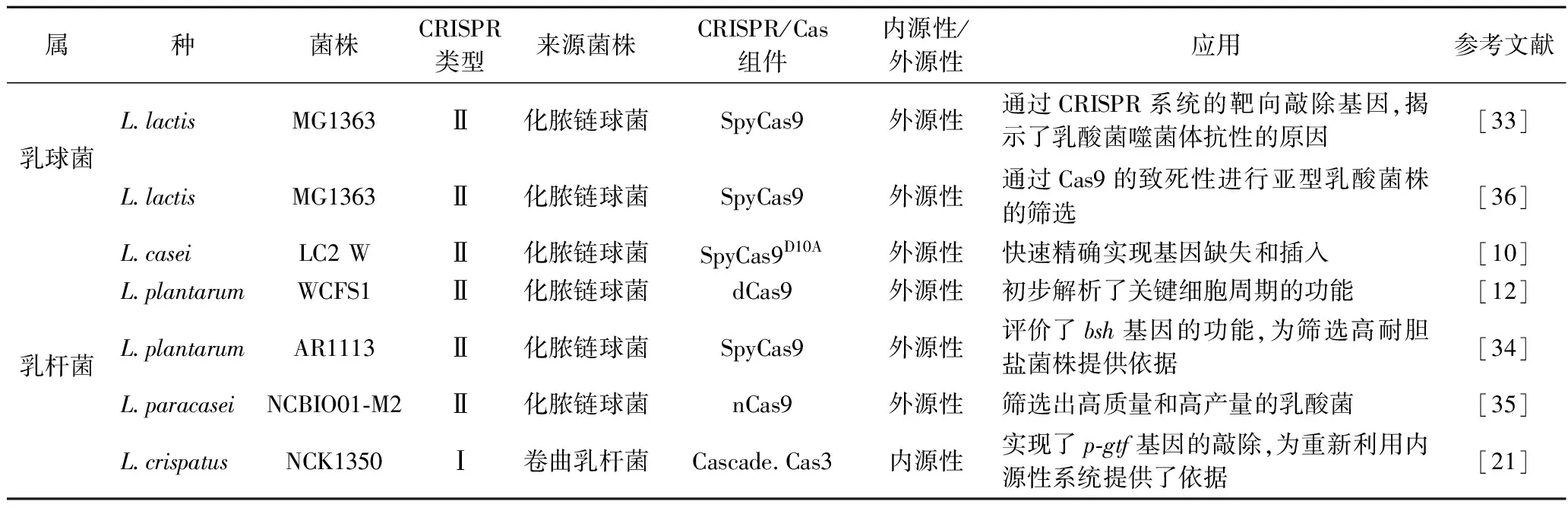
表3 CRISPR介导的基因编辑Table 3 CRISPR mediated gene editing
bacillussanfranciscensis)菌株。SCALTRITI等[39]分析了25株瑞士乳杆菌CRISPR序列差异性,所有菌株均属Ⅰ型系统且发现一个新的crispr重复家族lhel3。应用这种方法为预测CRISPR的潜在功能以及演化方向提供了一定的见解(表4)。

表4 利用CRISPR序列分析Table 4 Interspecific classification using CRISPR system
4 展望
CRISPR-Cas系统作为一种强大的基因编辑技术,已广泛用于基因编辑、碱基编辑、转录调控、荧光成像等领域,为各个行业提供了一种新型生物技术工具,从最初的CRISPR免疫系统到基因编辑系统的开发及不断完善,但在乳酸菌基因编辑方面还不够成熟,虽然二型CRISPR系统在乳酸菌中得到广泛应用,但其存在一定的局限性,不同乳酸菌外源性Cas9质粒转化条件不同,需要进行转化条件的优化为接下来的操作打下基础。Cas9质粒转化后Cas9蛋白有时并不能与靶标基因进行精准的结合,导致其产生脱靶效应。STOUT等[41]将干扰质粒导入加氏乳杆菌筛选出靶向逃逸的Cas9,并在此基础上发现间隔子的缺失是Cas9靶向逃逸的原因之一。Cas9靶向双链断裂时乳酸菌自身缺陷所带来的基因毒性等副作用和sgRNA的表达量不足影响后续的基因组编辑,同时因其在乳酸菌中不同物种之间对双键断裂的致死率差异较大,对基因组应用的范围得不到扩展,LEENAY等[42]证实了植物乳杆菌菌株之间基因编辑的差异性,在3种不同的植物乳杆菌菌株(WCFS1,WJL和NIZO2877)中,对SpyCas9编辑的2种不同方法进行了直接比较,发现在多个植物乳杆菌菌株中编辑同一位点并不总是成功。所以未来需要更多功能独特的新型菌株来进行CRISPR系统开发。
当然,这些问题是可以解决的,目前已经有了有效的应对方案,利用电转化的方式将质粒转入乳酸菌中大大提高了转化效率。针对Cas9系统的脱靶效应,研究者可改变Cas9蛋白的特性,使其靶向目的基因更加精准,或通过设计更加具备兼容性的PAM序列来提高Cas9靶向的效率,该策略已在人体细胞中得以成功应用,扩展了Cas系统的靶向范围。通过Cas9切口酶大大降低了因双链断裂导致的致死性,通过利用不同强度的启动子优化基因编辑工具,PCR获得不同的启动子片段,利用无缝克隆的方法,替换原有的启动子提高了sgRNA的表达量。CRISPR-Cas9乳酸菌编辑技术已经成为研究人员的热点,从此系统的发现到可以自由对基因的改造实现了巨大的飞跃,在外源性CRISPR方面将会开发更加广谱且简便的编辑工具,对内源性CRISPR菌株的挖掘为以后新型乳酸菌菌株的开辟提供了可能性。而且在不久的将来将会出现更多的亚型和Cas蛋白的突变体并应用到乳酸菌中。
