超高效液相色谱-四极杆-飞行时间质谱法快速筛查香橼中180 种农药残留
2022-06-14刘光瑞朱仁愿王行智戚鹏飞
陈 婷, 张 文, 刘光瑞, 朱仁愿, 闫 君,王行智, 戚鹏飞, 陈 敏
(兰州市食品药品检验检测研究院,甘肃省种植中药材外源性污染物监测工程研究中心,兰州 730050)
香橼 Fructus Citri 为芸香科植物枸櫞Citrus medicaL. 或香圆Citrus wilsoniiTanaka 干燥成熟的果实,具有理气保肝、和胃活血、化痰止咳、温脾止泻等功效,被国家卫健委列为药食同源中药材[1]。香橼在种植过程中不可避免地会发生病虫草害,需要使用农药进行防治[2]。近年来,中药材残留农药超标的报道时有发生,国外对产自中国的菊花、枸杞、金银花等常用中药材进行抽样检测,发现90%的样品中被检测出农药残留[3-4],因此,建立香橼中农药多残留快速筛查方法对提升香橼质量安全检测水平十分必要。
在《中国药典》(2020 版) 中,农药的检测方法主要有液相色谱-串联质谱法和气相色谱-串联质谱法[5]。这些方法的灵敏度高,靶向性强,但分辨力低,只适合已知目标化合物的检测,对大范围非靶向化合物的筛查能力有限。Q-TOF 技术基于精确质量数、同位素分布和源内碎裂离子等条件对目标化合物进行定性测定,具有高通量、非靶向的优点,在无标准品的前提下,可快速、准确、高效地同时筛查复杂样品中大量目标化合物[6-11]。目前针对香橼中的农药残留检测尚未见报道。本文通过对比药典方法中固相萃取法和QuEChERS 法两种前处理方法,优化提取净化步骤,结合Q-TOF 快速筛查技术,建立了中药材香橼中180 种农药残留的高通量非靶向快速筛查方法,旨在为香橼中农药残留风险监测提供技术保障。
1 材料与方法
1.1 仪器与试剂
Agilent 1290 超高效液相色谱/6540 四极杆-飞行时间质谱仪(美国安捷伦公司) ,配有双电喷雾离子源(dual Jet Stream ESI); KS501 摇床和RV10旋转蒸发仪 (德国IKA 公司);Centrifuge 5810R高速离心机 (德国Eppendorf 公司);VORTEX-5涡旋混匀器(海门市其林贝尔仪器制造有限公司);EVA50A 氮吹仪 (中国北京普立泰科仪器有限公司);Milli-Q 超纯水机 (美国Millipore 公司);ME204/02 电子天平 (美国梅特勒公司) 。
180 种农药标准品 (纯度≥95 %,德国Dr Ehrenstorfer 公司);乙腈、甲醇、甲苯、甲酸和乙酸铵 (色谱纯,德国Merck 公司);无水硫酸镁、无水乙酸钠、N-丙基乙二胺 (PSA)、十八烷基硅烷键合硅胶 (C18)、硅胶、石墨化碳黑 (GCB) (分析纯,中国天津博纳艾杰尔科技有限公司);试验用水为Milli-Q 超纯水。
1.2 样品前处理
香橼采集自药材市场,粉碎后过50 目筛(355 ±13 μm)。准确称取3 g 样品 (精确至0.01 g) 于50 mL离心管中,加入15 mL 超纯水,涡旋使药粉充分浸润,放置30 min;加入15 mL 酸化乙腈 (含体积分数为1 %乙酸的乙腈溶液) ,涡旋混匀,置振荡器上剧烈振荡5 min;加入无水硫酸镁和无水乙酸钠混合粉末7.5 g,立即摇散,再置振荡器上剧烈振荡5 min,于4000 r/min 下离心5 min;取9 mL上清液,置于预先装有净化材料的分散固相萃取净化管 (无水硫酸镁900 mg、PSA 300 mg、C18300 mg、硅胶300 mg 和GCB 90 mg) 中,涡旋混匀,置振荡器上剧烈振荡5 min,于4000 r/min 下离心5 min;取5 mL 上清液,置氮吹仪上于40 ℃水浴浓缩至近干,用V(乙腈) :V(水) = 3 : 2 溶液稀释并定容至1.0 mL,涡旋混匀,过0.22 μm 微孔滤膜,制得空白基质溶液,待测定。
1.3 检测条件
色谱条件:Agilent ZORBAX SB-C18色谱柱(2.1 mm × 100 mm,3.5 μm);流动相A 相为0.1%甲酸-5 mmol/L 乙酸铵水溶液,B 相为乙腈-0.1%甲酸溶液。梯度洗脱程序:0 min,90% A;1 min,70%A;>1~15 min,40%A;>15~23 min,10%A;23.01 min ,90%A, 保持4 min。流速0.4 mL/min;柱温 40 ℃;进样量2 μL。
质谱条件:电喷雾电离源正离子模式(ESI+);干燥气温度325 ℃;干燥气流速10 L/min;雾化器压力3.1 × 105Pa;毛细管电压4000 V;喷嘴电压500 V;鞘气温度325 ℃;鞘气流速11 L/min;全扫描(TOF-Scan) 模式;全扫描范围:m/z50~1000;目标化合物二级碎片离子扫描模式(Targeted MS/MS)m/z50~1000,在Spectral Parameters 项下Targeted List 中设置目标母离子MS/MS 碰撞能量分别为5、15、25、35 和45 V,180 种农药及其质谱信息见表1。
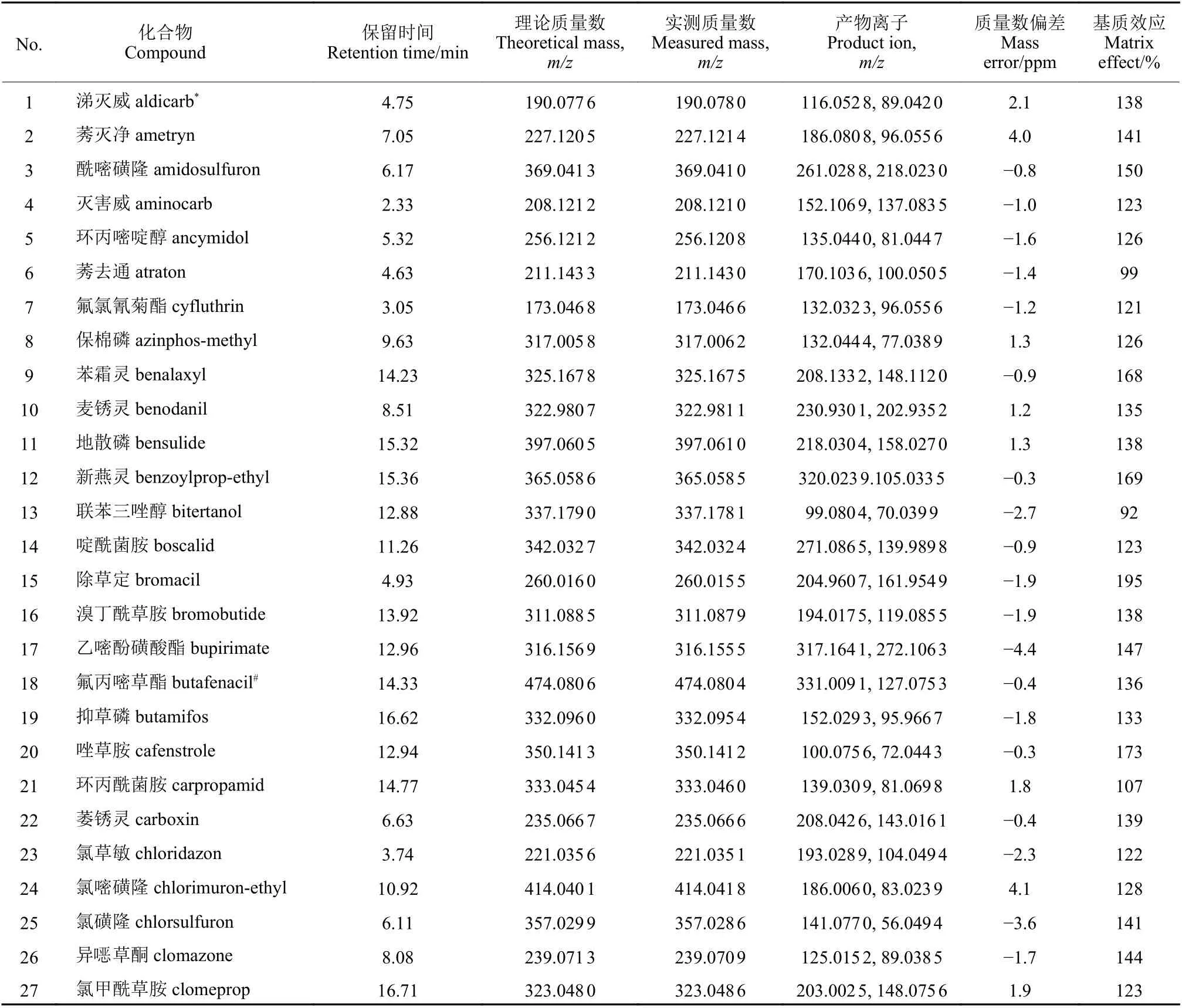
表1 180 种农药的保留时间、基质效应及质谱信息Table 1 Retention times,matrix effects and mass parameters of 180 pesticides
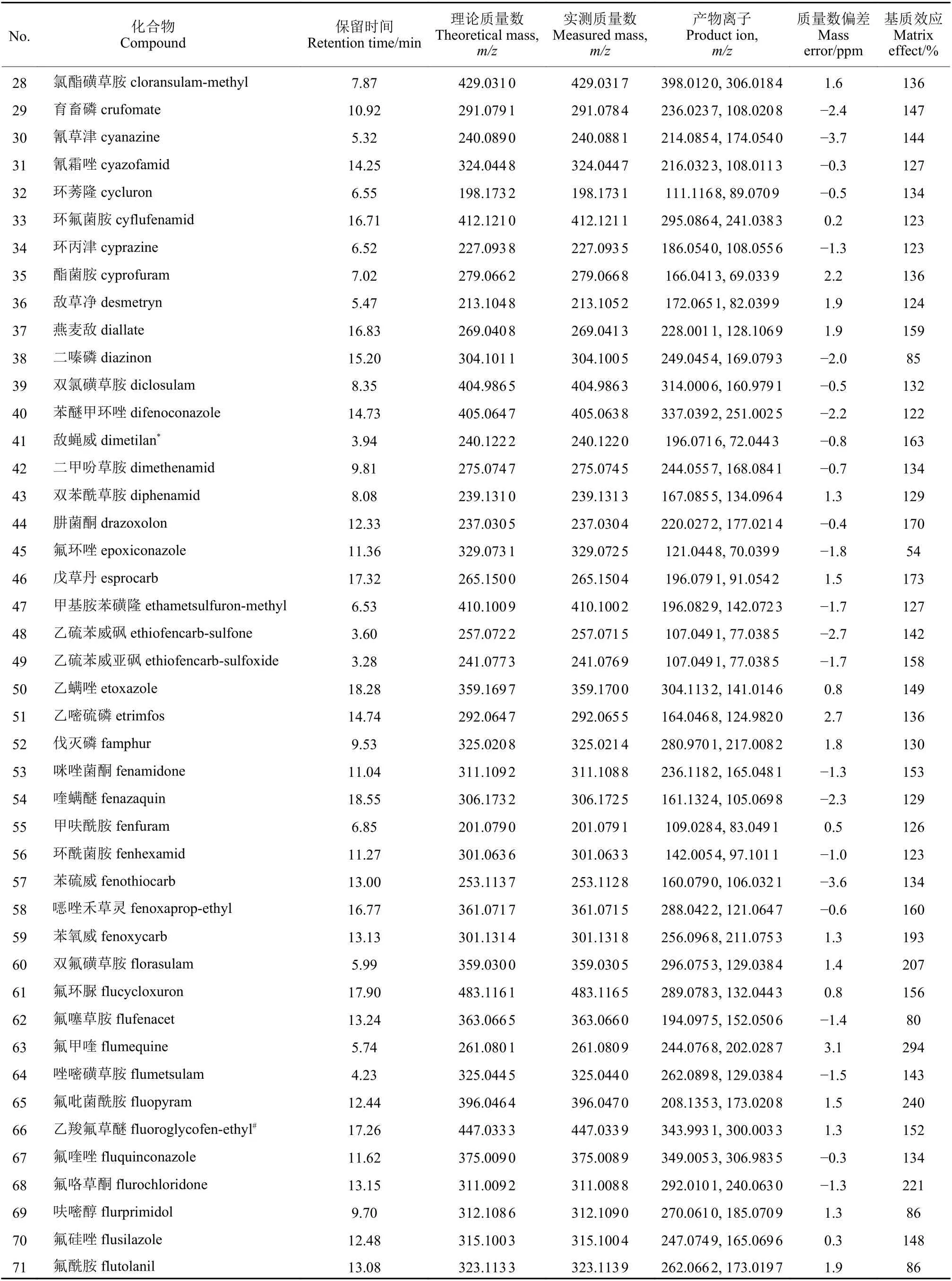
续表1Table 1 (Continued)

续表1Table 1 (Continued)

续表1Table 1 (Continued)

续表1Table 1 (Continued)
1.4 标准溶液的配制及标准曲线的绘制
1.4.1 标准储备液的配制 称取180 种农药标准品各25 mg,分别置于25 mL 容量瓶中,根据标准品的性质选择丙酮、甲醇等溶剂溶解并定容至刻度,配制成质量浓度为1000 mg/L 的单一农药标准储备液,于 −18℃避光保存。
1.4.2 混合标准液的配制 准确移取一定体积的单一农药标准储备液于25 mL 容量瓶中,用甲醇稀释并定容至刻度,配制成质量浓度为1 mg/L 的混合标准液,于4 ℃避光保存。
1.4.3 基质匹配混合标准工作液配制 量取农药混合标准液,用空白基质溶液稀释,配制成质量浓度分别为0.002、0.005、0.01、0.02、0.05 和0.1 mg/L 的系列基质匹配混合农药标准工作液,于4 ℃避光保存。
取基质匹配混合农药标准工作液按1.3 节的条件测定,以化合物浓度(x) 为横坐标,离子对峰面积(y) 为纵坐标,绘制标准曲线。
1.5 数据库的建立
按优化的色谱条件进样,对目标化合物进行TOF-Scan 全扫描,利用定性分析软件Mass Hunter Qualitative Analysis 对结果进行分析,获得目标物的保留时间、分子式、母离子、质量偏差及单一同位素准分子离子的质子化方式等信息,建立目标化合物一级精确质量数据库。利用targeted MS/MS 采集模式,在不同碰撞能量下采集一级数据库中目标物精确质量的母离子,获得碎片离子质谱图,导入PCDL 软件,并编辑对应目标化合物二级离子信息,建立适用于多农药残留非靶向筛查的二级离子质谱库。
2 结果与讨论
2.1 净化剂的优化
QuEChERS 方法是利用吸附剂填料与基质中的杂质相互作用,通过分散固相萃取(DSPE) 达到除杂净化的目的。本研究选用15 种具有代表性的农药作为前处理方法开发的研究对象,其中按化合物种类包括三嗪类农药 (如莠灭净)、酰胺类农药 (如苯霜灵)、氨基甲酸酯类农药 (如戊草丹)、有机磷类农药 (如喹硫磷) 和嘧啶类农药 (如嘧菌胺) 等农药类型;按功能分类包括除草剂、杀菌剂、杀虫剂和杀螨剂等,从化合物种类和功能性上均具有代表性,且这15 种农药性质稳定,可有效提高方法优化的可靠性。针对香橼基质提取物中含有的糖类、有机酸、色素、脂类等物质,考察了3 组净化剂组合的净化效果:组合1 为:PSA 150 mg,GCB 15 mg,MgSO4900 mg;组合2 为:PSA 150 mg,C18150 mg,MgSO4900 mg;组合3 为:PSA 300 mg,C18300 mg,MgSO4900 mg,GCB 90 mg,硅胶300 mg。结果 (图1) 表明:组合3 处理后的样品平均回收率为102%,组合1 的为96%,组合2 的为108%,其中组合3 处理后回收率在70%~120% 范围的农药数占93%,组合1 为80%,组合2 为67%。故最终选择组合3 作为净化吸附剂。

图1 不同净化方式对香橼中15 种农药回收率的影响Fig. 1 Effects of cleanup sorbent combinations on recoveries of 15 pesticides in Fructus Citri
2.2 净化方法的比较
对QuEChERS 法和固相萃取法 (SPE) 两个前处理方法进行了对比研究。结果 (图2) 表明:固相萃取法净化所得样品上机液呈淡黄色,且糖类及挥发油类净化不完全,15 种化合物的平均回收率为84%,较QuEChERS 法结果偏低,可能是由于基质中杂质干扰的原因。最终选择QuEChERS净化方式。
2.3 筛查方法的优化
样品按优化后的方法进行前处理,采用TOFScan 模式在不同碰撞能量下进行全扫描,通过Mass Hunter Qualitative Analysis 中的 “Find Compounds by Formula” 算法自动分析采集的原始数据,提取其离子流色谱图 (EICs) 中每个可能的母离子及其在指定色谱保留时间范围内的碎片离子信息,分析其中可能存在的化合物,通过与已建立的180种农药PCDL 数据库进行自动检索,列出与数据库中目标化合物的精确质量数偏差 ± 10 ppm、保留时间偏差 ± 0.25 min、且加权匹配得分≥70 的疑似阳性结果,将疑似结果与二级质谱库进行匹配,利用碎片离子及其丰度比二次确证,若匹配得分大于70,则完成目标化合物的筛查识别与确证。如表2 所示,利用上述条件对1 批香橼样品进行筛查,结果共筛查出9 种化合物,其中氟甲喹、西玛津、灭害威和氟环唑未检测到其碎片离子信息,得分均小于70,则被判定为假阳性结果。
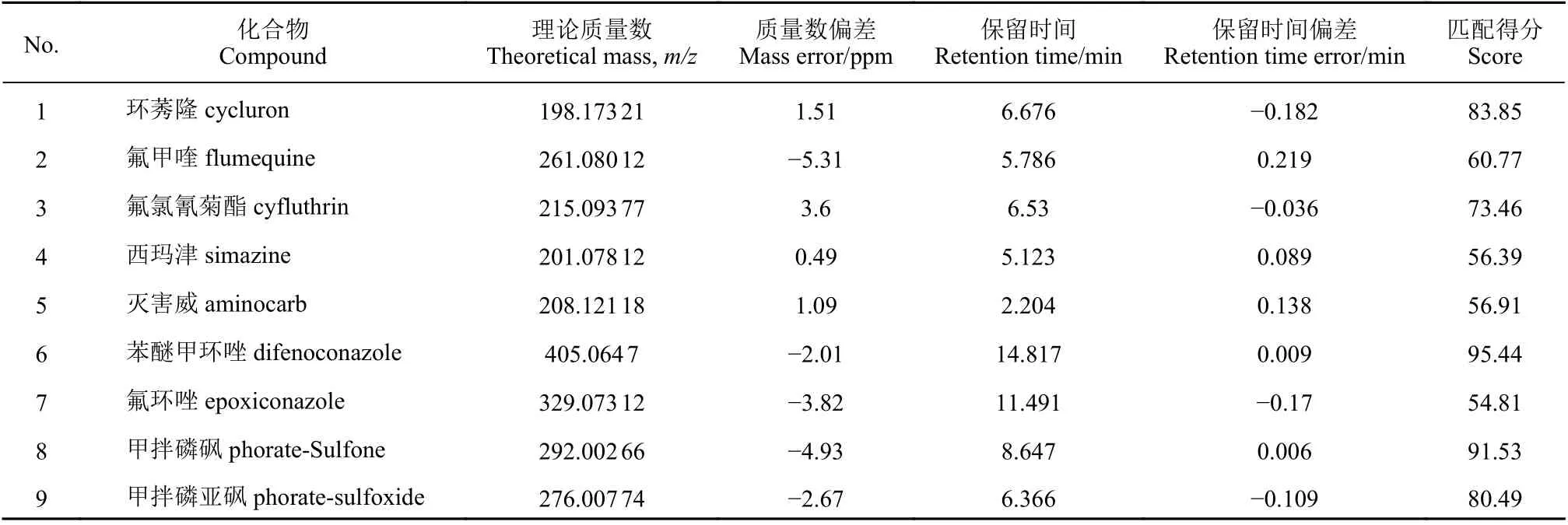
表2 香橼样品定性筛查结果Table 2 Qualitative screening results of Fructus Citri sample
采用上述自动筛查流程,对空白基质添加样品依据各个化合物的保留时间、化合物离子信息库,结合筛查参数进行非靶向定性匹配。为提高筛查效率,保证筛查结果的准确性,需对质量数提取窗口、保留时间窗口及峰面积阈值等筛查检索参数进行优化。
2.3.1 精确质量数提取窗口 精确质量偏差是理论值与实际测定值之间的差异,因此,精确质量数提取窗口也是化合物准确定性的重要依据之一。设置了 ±10 ppm、±30 ppm、±100 ppm 3 组质量数提取窗口。 结果显示:在一定的保留时间窗口及绝对峰面积范围内,质量数提取窗口为 ±10 ppm时,同一添加水平的化合物中未检出的化合物数量明显多于其他两组,以添加水平为0.05 mg/kg的乙螨唑为例,结果见图3:3 组质量数提取窗口提取m/z= 304.11322 的离子峰型差异较大,在± 100 ppm 时,干扰峰的出现会造成假阳性化合物的出现,考虑到虽然较低的质量数偏差可有效降低背景干扰,但同时也增大了假阴性的可能性,而较大的质量数偏差却降低了定性筛查质量准确度,综合考虑,将精确质量数窗口设置为 ± 30 ppm。

图3 不同精确质量数窗口下提取乙螨唑 (m/z = 304.113 22) 离子的结果Fig. 3 Extracted ion chromatogram of etoxazole at different mass width (m/z = 304.113 22)
2.3.2 保留时间窗口 精确的保留时间可有效区分同分异构体和同量异位素化合物,从而提高筛查分析的效率。优化了不同保留时间窗口(0.15、0.25、0.35、0.45 和0.55 min) 对检索结果的影响。结果发现:当保留时间窗口低于0.35 min时,多数化合物无法被检索出,造成假阴性结果增多,但随着时间窗口的增大,同一时间窗口内检出多个干扰色谱峰,又会造成化合物假阳性筛查数量增多。综合考虑,选择0.35 min 为保留时间窗口。
2.3.3 绝对峰面积阈值 准确的定量结果要求化合物色谱峰峰形好,分离度高,在定性匹配时避免提取背景和干扰离子。对比不同峰面积阈值(1000、3000、5000 和10000) 的结果显示,过大的峰面积阈值会增加假阴性样品的数量,对于响应值较低的化合物易出现漏筛现象。而当峰面积阈值小于3000 时,筛查结果中假阳性化合物数量也会明显增多,因此选择绝对峰面积3000 和相对峰面积≥ 0.1%作为提取未知物色谱峰的阈值。
2.4 基质效应
基质效应(Me) 是目标化合物离子化过程中受到基质共提物影响,导致化合物响应信号的增强或抑制。依据不同化合物的响应值高低,配制了0.005~0.1 mg/kg 范围的纯溶剂标准溶液及基质标准溶液,通过对比目标物在纯溶剂与基质溶液中的峰面积比值,判断基质效应的强弱。Me< 80%为基质抑制效应,80%
2.5 方法的线性范围、筛查限及定量限
结果表明:除涕灭威、氟甲喹和磺酰草吡唑外,其他目标化合物在0.005~1.0 mg/L 质量浓度范围内线性关系良好,相关系数 (r) 均≥ 0.99。
参照欧盟标准SANTE/12682/2019[12],筛查限(SDLs) 是指同一农药在95%的添加样品中均被检出的最低含量。在分析的180 种农药中,其中有99 种 (55.0%) 化合物能够在0.005 mg/kg 的含量水平中被检出,则SDLs 均 ≤ 0.005 mg/kg,有67种 (37.2%) 和8 种 (4.4%) 农药分别在0.01 mg/kg和0.02 mg/kg 的含量水平中被检出,对应的SDLs 分别为0.005~0.01 mg/kg (含0.01 mg/kg) 和0.01~0.02 mg/kg (含0.02 mg/kg) ,仅保棉磷、异柳磷、菜草畏、氧特丁硫磷砜、磺酰草吡唑和涕灭威6 种农药 (3.3%) 因响应较差而未能均在95%的添加样品中检出,由此推断这些农药在该方法中的SDLs > 0.05 mg/kg。通过对比欧盟MRL 标准中的限量值范围,本方法所建立的SDL 值均能满足中国、欧盟等国家或组织对香橼中大多数农药的最大残留限量检测要求。
按信噪比大于10 对应的添加水平作为定量限(LOQ),则有44 种 (24.4%) 化合物的LOQ 为0.005 mg/kg,104 种 (57.8%) 化合物的LOQ 为0.01 mg/kg,26 种 (14.4%) 化合物的LOQ 为0.02 mg/kg,6 种 (3.3%) 化合物的LOQ 为0.05 mg/kg。
2.6 方法的正确度及精密度
参照《中国药典》(2020 版) 通则中33 种禁用农药的定量限在0.01~0.2 mg/kg 之间[5],验证0.02、0.05 和0.1 mg/kg 3 个添加水平下180 种农药的正确度及精密度,每个添加水平重复5 次。结果显示:在3 个添加水平下180 种农药的平均回收率分别为58%~129%,68%~131%和66%~132%,其中87.2%的农药平均回收率在70%~120%之间,且RSD 为0.7%~18%。由于净化剂GCB 对其具有较强的吸附作用 (如喹氧灵),或是酰嘧磺隆等磺酰脲类农药为弱酸性化合物,在酸性较高的香橼基质中易发生降解,造成回收率降低 (低于70%);回收率高于120%的农药,可能是由于前处理过程中杂质净化不完全,对农药的提取率造成了干扰,导致回收率较高。
2.7 实际样品的检测
利用本方法对市售的50 批香橼样品中180 种农药残留进行了检测。结果显示:有7 份样品检出农药13 种,检出率为7.2%。所有检出农药共流出离子及其丰度比与谱库信息相一致,对比标准品数据库,参考碎片离子质谱图,匹配度均大于70%。以甲拌磷亚砜为例,实际样品检出农药的色谱及碎片离子图如图4 所示。采用基质匹配校准曲线对筛查出的农药进行定量分析,同时采用超高效液相色谱质谱联用仪 (UPLC-MS/MS) 对筛查和定量结果进行比对,结果如表3 所示:UPLC-Q-TOF/MS 法和UPLC-MS/MS 法测定结果相近 (相对误差< 23.7%) ,其中,甲拌磷(含甲拌磷砜、甲拌磷亚砜)含量分别为0.2595 和0.2314 mg/kg,超出其《中国药典》 2020 年版) 中的限量规定(0.02 mg/kg) ;氟啶虫胺腈含量分别为0.5062 和0.4985 mg/kg,超出其欧盟最大残留限量标准(0.4 mg/kg);其他检出农药含量均未超过欧盟限量标准。
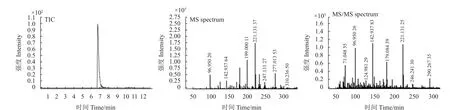
图4 检出样品中甲拌磷亚砜的色谱图和质谱图Fig. 4 Chromatogram and mass spectra of phorate sulfoxide in positive sample

表3 实际样品检测结果Table 3 Sample test results
3 结论
本文建立了香橼中180 种农药的QuEChERS结合超高效液相色谱-四极杆-飞行时间质谱检测方法,通过优化前处理条件及飞行时间质谱筛查过程中的精确质量数提取窗口、保留时间窗口及绝对峰面积阈值等参数,利用建立的精确质量数据库及高分辨碎片离子谱图库,实现了香橼中多种农药残留的快速非靶向筛查和准确定量。与传统的液相色谱-串联质谱法相比,该法具有高通量、高效率及高灵敏度等优点,筛查无假阳性,而且定量结果准确可靠,在中药材非靶向目标化合物和高通量多农残快速筛查方面具有较高的应用价值。
