基于WGCNA的棉花子叶抗冷相关共表达模块鉴定
2022-06-14周雨青杨永飞葛常伟沈倩张思平刘绍东马慧娟陈静刘瑞华李士丛赵新华李存东庞朝友
周雨青, 杨永飞, 葛常伟, 沈倩, 张思平, 刘绍东, 马慧娟,陈静, 刘瑞华, 李士丛, 赵新华, 李存东, 庞朝友*
(1.河北农业大学农学院,棉花生物学国家重点实验室河北基地,河北 保定 071001;2.中国农业科学院棉花研究所,棉花生物学国家重点实验室,河南 安阳 455000)
棉花是一种喜温作物,起源于热带和亚热带地区,近年来,我国主要棉区逐步“西进、东移、北上”[1],其栽培范围已逐步扩展至较冷区域。新疆植棉面积较大,该地区属于典型的大陆性气候,春季经常发生“倒春寒”,因而在播种出苗期及苗期常常遭受低温的危害,其中苗期冷害发生较频繁,大幅度降温、下霜并伴随降水的强烈灾害性天气对棉苗生长危害极大[2],每年因烂种、烂芽、死苗或晚发而导致部分棉田重播[3],严重影响了我国棉花的产量及纤维品质。因此,揭示棉花苗期抗冷机制和培育抗冷棉花品种具有重要意义。
加权基因表达网络分析(weighted gene coexpression network analysis,WGCNA)是基于大样本转录组数据的生物信息学分析方法,其首先假定基因网络服从无尺度分布,将表达模式相似的基因聚类形成不同的模块,分析模块与特定性状或表型之间的关联性,通过基因聚类和关联分析结果构建共表达调控网络。位于调控网络中心的基因被称为核心基因(hub gene),这类基因通常是关键的调控基因,值得深入挖掘和分析[4]。在棉花研究中,巨飞燕等[5]利用2个株型结构差异显著的棉花品种的18份不同长度果枝节间转录组数据构建共表达网络,鉴定到与棉花果枝节间伸长相关的特异性模块,并发现植物激素信号转导通路中的JAZ基因为该模块的枢纽基因。傅明川等[6]利用21份黄萎病菌侵染不同时间点的海岛棉幼苗根尖转录组数据构建共表达网络,鉴定到5个与抗黄萎病相关特异性模块,并挖掘出了网络中的核心基因。在抗冷研究中,李旭凯等[7]利用47份正常水稻组织转录组数据,通过冷胁迫、干旱胁迫、盐胁迫不同的处理方式,使用WGCNA方法挖掘到2 599个与3种胁迫都相关的基因,并预测出25个抗逆相关的关键基因,为水稻的综合抗逆能力等研究提供了新思路。秦梦凡等[8]利用2个抗冻响应有差别的甘蓝型油菜材料经不同低温处理的36个转录组数据,鉴定到了共同响应冻害和耐寒的特异性模块,并对其调控机制进行了分析,为油菜的耐寒调控机制研究提供重要的参考依据。而关于棉花抗冷研究的WGCNA分析未见报道。
本研究以4℃低温处理不同时间的棉花子叶转录组数据为材料,对其进行差异表达分析;通过构建加权基因共表达网络划分基因模块,筛选出抗冷相关的特异性模块;经GO富集分析探究模块功能,根据基因在相应网络中的连通性鉴定出模块内的核心基因。本研究可为进一步理解棉花低温冷害的分子机制提供理论基础,并为棉花抗冷育种提供新的基因资源。
1 材料与方法
1.1 试验材料及处理
试验材料新陆中16为抗冷品种(cold tolerance,CT),新陆中 32为冷敏感品种(cold sensitivity,CS),均由中国农业科学院棉花研究所种质资源中期库提供。棉花材料先于28℃光照培养室进行育苗(光照16 h,黑暗8 h),采用营养土与蛭石体积3∶1的混合基质培养,并保证水分充足,待子叶平展时将材料放入4℃冷室处理0、1、3、6、9、12 h,选取长势一致的棉苗对其子叶进行取样,每个时期3次重复,每次重复子叶数量为8~10片,共36个样本,对其进行转录组测序,得到各样本的基因表达量数据,使用FPKM(fragments per kilobase of exon per million fragments mapped)值来衡量基因的表达水平[9]。新陆中16和新陆中32的测序样品组设为CT和CS,根据冷处理时间0、1、3、6、9、12 h,新陆中16处理组命名为 CT0、CT1、CT3、CT6、CT9、CT12,新陆中 32 处理组命名为CS0、CS1、CS3、CS6、CS9、CS12。
1.2 权重基因共表达网络构建
对选用的基因集进行筛选过滤,需同时满足以下条件:基因在样本中的表达量均值要大于1,超过一半的样本表达量大于0,变异系数大于0.2。去掉不符合条件的低质量基因,提高网络构建的精度。利用R软件中的WGCNA(v1.47)包计算权重值,完成权重基因共表达网络的构建。首先根据所有基因的表达量对所有样本进行聚类,以分析样本关系。根据无尺度网络原则确定软阈值(soft thresholding power),取相关系数达到平台期(或大于0.8)时最小power值作为后续分析参数,同时统计在不同power值下基因平均连通性的变化。采用动态切割法(dynamic tree cut)对基因进行聚类及模块划分。根据基因间表达量的相关性构建基因聚类树,并根据基因间的聚类关系划分基因模块,然后根据模块特征值的相似度合并表达模式相近的模块。设定模块最少基因数为50,相似模块合并阈值为0.8,将模块基因在各个样本中的表达模式用模块特征值来展示,并绘制样本表达模式热图。通过模块特征向量基因(module eigengene,ME)分析确定与抗冷显著相关的特异性模块,后续选择相应的模块进行深入研究。
1.3 核心基因的筛选和功能分析
为进一步挖掘特异性模块的功能,对特异性模块基因进行GO功能分析。通过GO数据库(http://www.geneontology.org/)进行GO分类,结果显示目标模块基因可以归为分子功能(molecular function)、生物过程(biological process)和细胞组分(cellular component)3个大类。经过多重检验校正后,以P值<0.05为阈值,满足此条件的定义为在基因中显著富集的GO term。
为获得特异性模块中的核心基因,利用Cytoscape3.7.1软件对基因互作网络进行可视化处理,根据模块中基因的连通性(connectivity)及转录组数据中基因的FPKM值,筛选出连通性高且表达量高的基因,将这些基因作为该模块中的核心基因。在这些网络中,每个节点代表1个基因,处于连线两端的基因通常被认为具有相同的生物功能。基因调控关系网络图能准确筛选与核心基因存在调控关系的候选基因,并利用已知基因的功能预测未知基因功能。
1.4 核心基因的qRT-PCR验证
将用于转录组测序的备份RNA反转录,利用SYBR PremixEx Taq(DRR041A)荧光定量试剂盒对筛选得到的核心基因进行qRT-PCR(quantitative real-time polymerase chain reaction)验证,检测核心基因在CT和CS中对冷胁迫的响应情况。通过网站qPrimerDB(https://biodb.swu.edu.cn/qprimerdb/)设计qRT-PCR引物(表1)。内参基因为GhUBQ7(Gh_A11G0969),每个反应设置3个重复。用2-ΔΔCT方法[10]计算基因的相对表达量。

表1 qRT-PCR所用基因引物序列Table 1 Specific primers for the selected genes
2 结果与分析
2.1 构建权重基因共表达网络
2.1.1 软阈值确定 从图1可以看出,当软阈值β=8时,无尺度网络拟合指数R2>0.8,平均连通性趋近于0,表明用此值进行幂处理可以得到符合要求的无尺度网络,因此选择β=8构建无尺度网络。

图1 软阈值的确定Fig.1 Determination of soft threshold power
2.1.2 基因共表达网络中基因的聚类 根据基因间表达量的相关性构建聚类树,采用动态切割法将产生的聚类树切割,把表达模式相似的基因合并在同一分支上,每个分支代表1个共表达模块,根据模块相似度(0.8)对表达模式相似的模块合并后进行模块划分,最终获得9个共表达模块(图2),每一种颜色代表一个模块,灰色(Grey)模块代表无法归入任何一个模块的基因。蓝色(Blue)模块包含的基因数量最多,为16 025个,其次是棕色(Brown)模块,包含1 893个基因,黄色(Yellow)模块包含1 818个基因,灰色(Grey)模块包含655个基因,黑色(Black)模块包含616个基因,绿色(Green)模块包含489个基因,洋红色(Magenta)模块包含337个基因,粉色(Pink)模块包含184个基因,黄绿色(Greenyellow)模块所含基因最少,为66个(图3)。
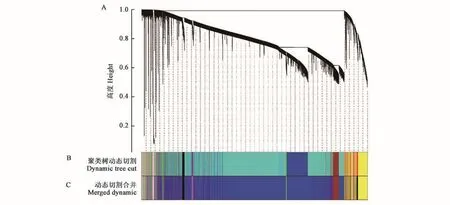
图2 基因聚类树和模块划分Fig.2 Gene cluster dendrograms and module division

图3 共表达模块中基因数量分布Fig.3 Distribution of number of genes in co-expression modules
2.1.3 基因共表达网络中模块与样本的关联分析 将所获模块与样本进行关联分析,得到9个与不同品种和处理时间相关联的模块,部分模块与品种和处理时间高度关联。例如,Brown模块的基因表达模式随着低温处理时间的增加在2个品种中均存在由负到正的趋势,与低温处理9和12 h时间点正相关,与低温处理0、1、3和6 h时间点负相关;Blue模块的基因表达模式随着低温处理时间的增加在两个品种中存在由正到负的趋势,与低温处理0、1和3 h时间点正相关,与低温处理6、9和12 h时间点负相关(图4)。

图4 基因共表达网络模块与不同样本的关联热图Fig.4 Association analysis of gene co-expression network modules with different samples
模块中具有代表性的基因即为特征向量基因(ME),它们之间的相关性越高,其所在模块的相关性也就越高,通过ME相关性分析发现,Blue模块与Brown模块中ME之间的相关性达到-0.86(图5)。因此将这两个模块作为抗冷相关特异性模块进行深入研究,挖掘模块中的核心基因。

图5 不同模块两两之间ME的相关性Fig.5 ME correlation between different modules
2.2 核心基因的筛选和功能分析
对Blue和Brown模块进行GO功能富集分析发现,这2个模块都可以显著富集到生物学过程(P)、分子功能(F)以及细胞组分(C)的若干GO通路(表2)。在Blue模块中筛选出了5个核心基因(图6),Brown模块中筛选出了5个核心基因(图7),利用棉花基因数据库(https://cottonfgd.org/)和NCBI数据库(https://www.ncbi.nlm.nih.gov/)获取这些核心基因的功能信息,并借助TAIR数据库(https://www.arabidopsis.org/)注释这些核心基因在拟南芥中的同源基因的功能(表3),结合已有研究进展,本研究发现这些筛选出的核心基因基本都与冷胁迫相关,比如Blue模块中的C2H2型锌指蛋白Gh_A13G2112富集到刺激反应(GO:0050896)、几丁质反应(GO:0010200)、冷反应(GO:0009409)、温度刺激反应(GO:0009266)、胁迫反应(GO:0006950)等多个通路中;Brown模块中参与冷调节的基因Gh_D09G1773和Gh_A05G1 554均富集到刺激反应(GO:0050896)、几丁质反应(GO:0010200)、温度刺激反应(GO:0009266)、冷反应(GO:0009409)、外部刺激反应(GO:0009605)、内源性刺激反应(GO:0009719)等多个通路。
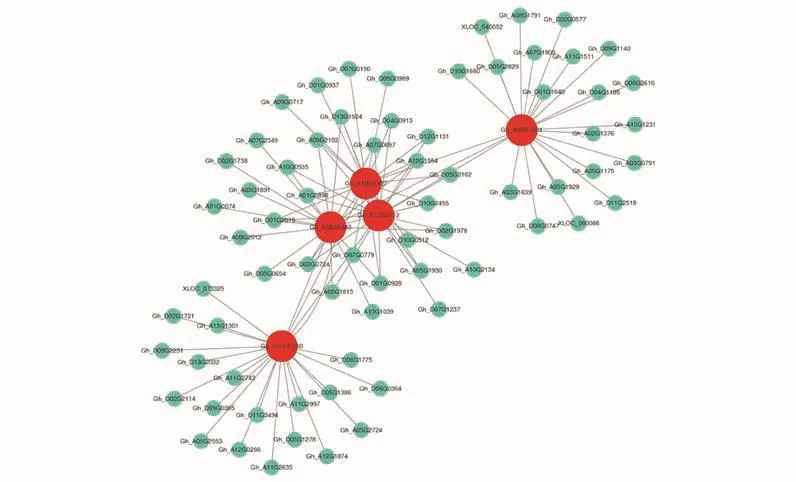
图6 Blue模块的基因共表达网络及其核心基因Fig.6 Gene co-expression network and hub genes in Blue module
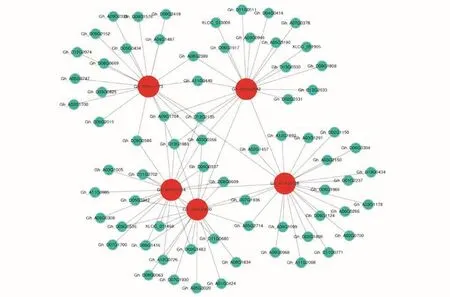
图7 Brown模块的基因共表达网络及其核心基因Fig.7 Gene co-expression network and hub genes in Brown module
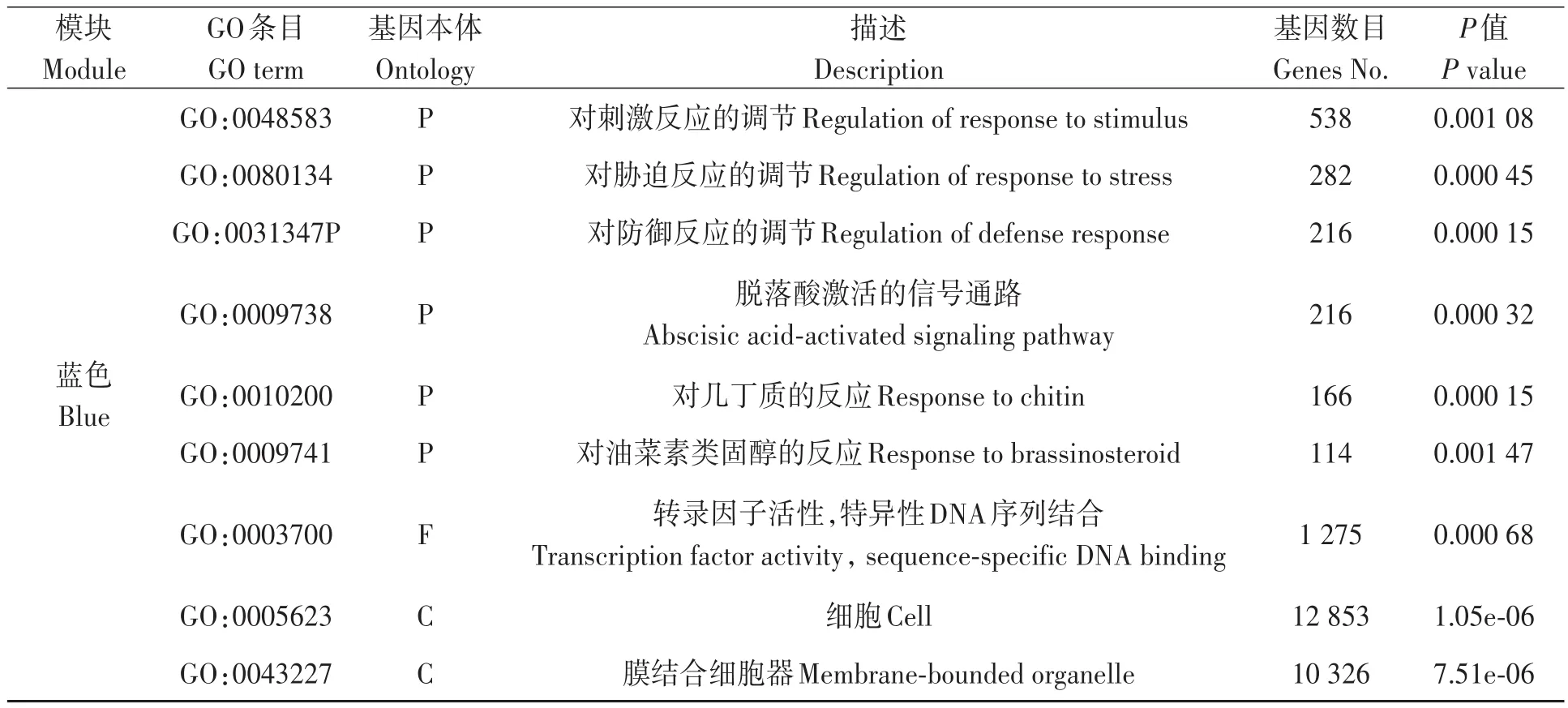
表2 特异性模块的部分GO富集分析结果Table 2 Partial GO enrichment analysis of target module

表2 特异性模块的部分GO富集分析结果Table 2 Partial GO enrichment analysis of target module 续表Continued
2.3 核心基因的RT-qPCR验证
从相关性最高的2个基因模块中筛选到10个可能与冷胁迫相关的核心基因(表3),对这10个基因进行qRT-PCR验证,最终基因表达量的变化趋势与转录组结果基本一致(图8和9),进一步证明了转录组测序结果的可靠性。这10个核心基因的表达量在2个抗冷性差异品种中随着低温处理时间的增加而增加,均呈现上调表达的趋势,验证了这些核心基因响应冷胁迫。
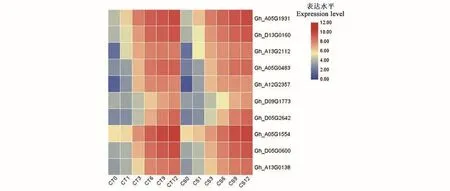
图8 核心基因的表达模式Fig.8 Expression pattern of hub genes
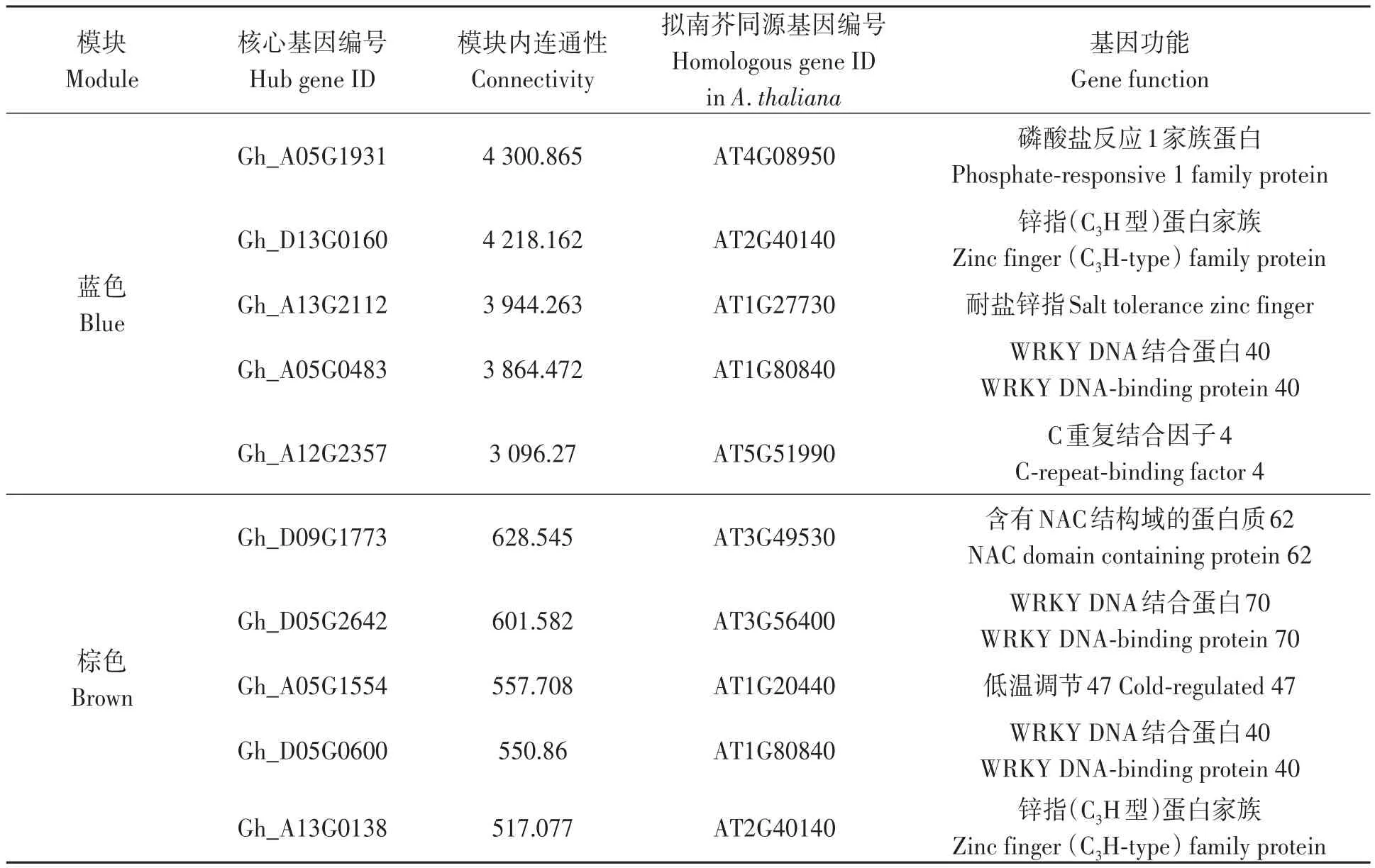
表3 目标模块中核心基因的功能注释Table 3 Annotation of hub genes in target module

图9 核心基因的RT-qPCR验证Fig.9 qRT-PCR validation of hub genes
3 讨论
本研究通过WGCNA富集到9个冷胁迫处理下与棉花品种和处理时间相关联的共表达模块,利用各个模块的特征向量基因对模块进行相关性分析,发现Brown和Blue模块间存在极显著的相关性且关联度最高,可作为子叶抗冷性状机理研究的目标模块。对这2个模块内的基因进行GO功能富集分析,均富集到了抗逆相关的具有生物学意义的调控途径。例如,Blue和Brown模块富集到转录因子活性(GO:0003700)、对胁迫的反应(GO:0006950)等通路,该结果与谢勇军[11]的研究结果一致。
对这2个模块的核心基因进行分析发现,核心基因里也有部分基因与冷胁迫相关,如Gh_A05G1931、 Gh_A13G2112、 Gh_A12G2357、Gh_D09G1773、Gh_A05G1554。筛选出的核心基因在模式植物拟南芥和其他作物(如玉米、水稻等)中有较多的研究,但在棉花中未见报道,可进行深入探究。Gh_A05G1931(AT4G08950)参与了对油菜素类固醇(brassinosteroids,BR)的反应。遏蓝菜(Thlaspi arvense)比其近缘植物拟南芥(Arabidopsis thaliana)或甘蓝型油菜(Brassicanapus)具有更高的抗冻性,原因可能是一些基因在表达水平和表达时间上存在差异,试验证明EXO(AT4G08950)基因在甘蓝型油菜和遏蓝菜中均显著上调表达,但在拟南芥中没有观察到变化,且在甘蓝型油菜中表达量的增加最为明显[12],说明EXO基因能增强遏蓝菜的抗冻性。玉米中,用油菜素类固醇进行浸种处理,可增加幼苗可溶性糖含量,减轻细胞膜伤害,提高脯氨酸含量,从而增强 植 物 对 低 温 的 抗 性[13]。 Gh_A13G2112(AT1G27730)属于C2H2型锌指蛋白家族。C2H2锌指蛋白含有ERF相关的两亲性抑制(EAR)结构域[14-16],在调节植物对非生物胁迫的防御反应中起着关键作用。ZAT10/STZ(AT1G27730)最初被鉴定为盐和冷反应蛋白[17]。ZAT10转录受脱落酸、干旱、氯化钠、低温和强光等多种非生物胁迫的高度诱导。过表达ZAT10的转基因植物表现出生长迟缓,同时它们表现出对多种非生物胁迫的耐受性增强,并且敲除该基因也可以增强对盐、渗透胁迫的耐受性[15,17-20]。此外,ZAT10为拟南芥丝裂原活化蛋白激酶(MAPKs或MPKs)的直接靶向底物,其活性可能通过MPK直接磷酸化来调节[21]。植物MPKs参与了对各种生物和非生物胁迫的胁迫耐受反应,如病原体感染、低温、低湿度、盐、伤害、紫外线、臭氧和重金属[22-27]。Gh_A12G2357(AT5G51990)是C-重复结合因子/脱水反应结合元件(CBF/DREB1)家族的成员。CBF基因家族主要包括4个成员:CBF1、CBF2、CBF3和CBF4[28]。冷处理下CBF1和CBF3基因的表达既不上调也不下调,但激活了CBF2和CBF4(AT5G51990)基因,尤其是低温胁迫下CBF4基因的表达高度上调[29]。CBF4过表达使转基因拟南芥更耐寒和耐旱[30]。Gh_D09G1773(AT3G49530)是植物特异转录因子NAC家族的成员,在发育过程和胁迫反应中均起作用[31]。研究发现,NAC062(AT3G49530)受冷诱导活化,进入细胞核调控抗病相关基因,提高植物抗病能力,冷害胁迫期间NAC062的活化可能由膜组成和流动性改变导致[32]。Gh_A05G1554(AT1G20440)属于脱水蛋白家族,与脱落酸途径有关,当其过表达时,植物具有耐寒性,是一种冷调节基因。脱水蛋白基因通常在植物脱水时表达,脱水可能是由干旱、渗透胁迫或低温引起的[33]。有研究表明,COR47(AT1G20440)是在低温下积累的主要脱氢酶(DHNs),有助于冷应激反应,在体外已实现COR47的离子和水结合、低温保护活性、类囊体膜结合和金属结合等功能,且COR47的过表达与低温条件下拟南芥抗冷性的提高有关[34]。
以上结果表明,利用WGCNA分析可挖掘到与目标性状的生物学意义高度关联的基因模块和核心基因,为挖掘目标基因提供新的研究思路,为解析复杂的农艺性状提供参考。本研究重点关注了Blue和Brown这2个相关性高的调控模块,其余的基因模块虽然没有被详细论述,但也可能包含与子叶抗冷相关的通路,可进一步挖掘其蕴含的生物学意义。
