平面五配位硅、锗XBe5H 6(X=Si,Ge)团簇
2022-06-10郭谨昌刘芳林
郭谨昌,刘芳林
(山西大学分子科学研究所,纳米团簇实验室,太原 030006)
1970年,Hoffmann等[1]提出了平面四配位碳概念及其稳定策略,开启了平面多配位碳化合物的研究.近50年来,作为非经典成键探针,平面四、五、六配位碳化合物备受关注,在理论设计、光谱表征等方面取得了积极进展[2~22].其中,最具有代表性的体系包含平面四配位碳(ptC)CA、平面五配位碳(ppC)CA、平面六配位碳(p6C)团簇及其衍生物[10~22].
硅和锗与碳为同一主族,成键与其具有类似性.在平面多配位碳化合物研究的基础上,将平面多配位模式由碳自然拓展至硅和锗.2000年,Wang等[23]采用光电子能谱实验与密度泛函理论相结合的方法确定了五原子Al4Si-和Al4Ge-团簇的全局极小结构,发现它们含有平面四配位硅/锗(ptSi/Ge)中心.2004年,Li等[24]在密度泛函理论水平上提出了平面多配位硅统一结构模式BnE2Si(CH)n(E=CH,BH,Si;n=2~5).这些扇形结构的BnE2Si(C2v),随着外围B原子个数的增加扇形不断扩张,最终成为完美的平面八配位硅SiB8分子轮.2005年,Li等[25]理论预测了金属烃稳定的ppSi/Ge M5H5X(M=Ag,Au,Pd,Pt;X=Si,Ge)团簇.2007年,Merino等[26]理论预测了系列平面多配位14族原子为中心的硼分子轮团簇.2008年,Liu等[27]理论设计了含准平面四配位硅的C58Si富勒烯衍生物.然而,这些结构完美的平面多配位硅团簇仅为体系势能面上的局域极小结构,很难被实验合成及表征.对于理论研究,设计的团簇为全局极小结构十分重要,因为它们最有可能被气相谱学实验表征.近年来,理论预测了一些平面四配位硅全局极小团簇,包括SiI,X3(X=Si,Ge;M=Cu,Li),C2M2X(M=Si,Al,Ga,In,Tl;X=Si,Ge,Sn,Pb),Li3SiAs2-,HSiX3(X=Al,Ga),Ca3SiAl-,Mg4Si2-,C2LiSi,Si3X2(X=Li,Na,K)等[28~32].与平面四配位硅体系相比,理论设计平面五、六配位硅全局极小团簇更为困难.最近,Cui等[33]理论预测了系列平面五配位硅团簇XMg4Y-(X=Si,Ge;Y=In,Tl)及SiMg3In2,它们代表首例平面五配位硅全局极小.我们的系列研究[17,34,35]表明,低电负性主族金属铍成键方式灵巧,为稳定平面多配位碳的优良配体,在外围适量H原子辅助下,可稳定平面多配位碳、硼及过渡金属原子.而采用类似方式稳定平面五配位硅、锗还未见报道.在铍氢稳定的系列平面多配位碳团簇研究的基础上,本文采用铍作为配体配位硅、锗中心,氢原子作为外围辅助原子,理论设计了含平面五配位硅、锗的XBe5H6(X=Si,Ge)团簇,其外围形成2个Be—H(2c-2e)键及4个Be—H—Be(3c-2e)键,中心的ppSi/Ge原子满足稳定的八隅律(或八电子规则),所在的XBe5单元具有2π/6σ双重芳香性.
1 理论方法
对XBe5H6(X=Si,Ge)团簇的全局极小结构搜索采用Coalescence Kick(CK)[36~38]程序,在PBE0/def2-SVP水平上对目标体系的单重态和三重态各搜索了2000个结构.对筛选得到的低能量结构,在PBE0/aug-cc-pVTZ水平上重新进行了结构优化[39,40],并进行了频率分析,以确保其为势能面上的真正极小结构.为了获得更为精准的相对能量,在CCSD(T)/aug-cc-pVTZ//PBE0/aug-cc-pVTZ水平上对前8个低能量结构进行了单点能计算[41,42].为了进一步验证方法的可靠性,作为对照,还在B3LYP/aug-cc-pVTZ水平上对前8个结构进行了优化[43,44],并在CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ水平上计算了单点能.为了评估XBe5H6(X=Si,Ge)团簇的动力学稳定性,在298 K温度下,将优化的XBe5H6(X=Si,Ge)全局极小结构作为初始结构,在PBE0/def2-SVP水平上对XBe5H6(X=Si,Ge)团簇进行了50 ps的玻恩-奥本海默分子动力学(BOMD)模拟[45].
在PBE0/aug-cc-pVTZ理论水平上,对XBe5H6(X=Si,Ge)团簇的全局极小结构进行了自然键轨道分析(NBO)[46],从而获得韦伯键级和电荷数据.正则轨道成分分析和适应性自然密度划分(AdNDP)采用Multiwfn程序完成[47,48].采用ADF程序[49],在PBE0/TZ2P//PBE0/aug-cc-pVTZ水平上对XBe5H6(X=Si,Ge)团簇进行了能量分解(EDA)[50]及化学价自然轨道(NOCV)[51]分析.为了定量分析体系的π和σ芳香性,在PBE0/aug-cc-pVTZ水平上对其特定位置的核独立化学位移(NICSs)[52]值进行了计算.本文所有电子结构及BOMD计算均采用Gaussian 09程序[53]完成.分子结构、正则分子轨道(CMOs)和适应性自然密度划分(AdNDP)分别采用CYLview,GaussView及Molekel程序[54,55]完成.
2 结果与讨论
2.1 结构及稳定性
图1给出了PBE0/aug-cc-pVTZ理论水平上XBe5H6(X=Si,Ge)团簇前8个低能量结构的优化构型,同时标注了CCSD(T)/aug-cc-pVTZ//PBE0/aug-cc-pVTZ水平上的相对能量.作为对照,括号中还给出了CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ水平上的相对能量.由于两种方法给出的能量次序一致,本文主要基于PBE0及CCSD(T)//PBE0的结果进行讨论.为了简便起见,用序号1,2分别表示XBe5H6(X=Si,Ge)团簇的全局极小结构,nB~nH(n=1,2)表示相应的低能量异构体.如图1所示,1和2整体呈完美的扇形结构,5个Be原子以半包围方式与中心的Si/Ge原子配位,外围4个H原子以桥基方式与2个相邻的Be原子键连,其余2个H原子以端基方式分别与末端的Be原子键连.最具竞争的异构体1B/2B能量分别比全局极小结构1,2高出9.57,17.62 kJ/mol.在这些低能量结构中,Be原子倾向与Si/Ge原子配位,而H原子倾向以桥基或端基方式与Be原子成键.仅在1E/1H/2H结构中,1个H原子直接与Si键连.除结构1,2外,其余结构中Si/Ge原子配位数为4或3.由图1相对能量可知,结构1和2为真正的平面五配位Si/Ge全局极小结构.表S1(见本文支持信息)给出了XBe5H6(X=Si,Ge)团簇前8个低能量结构的笛卡尔坐标.
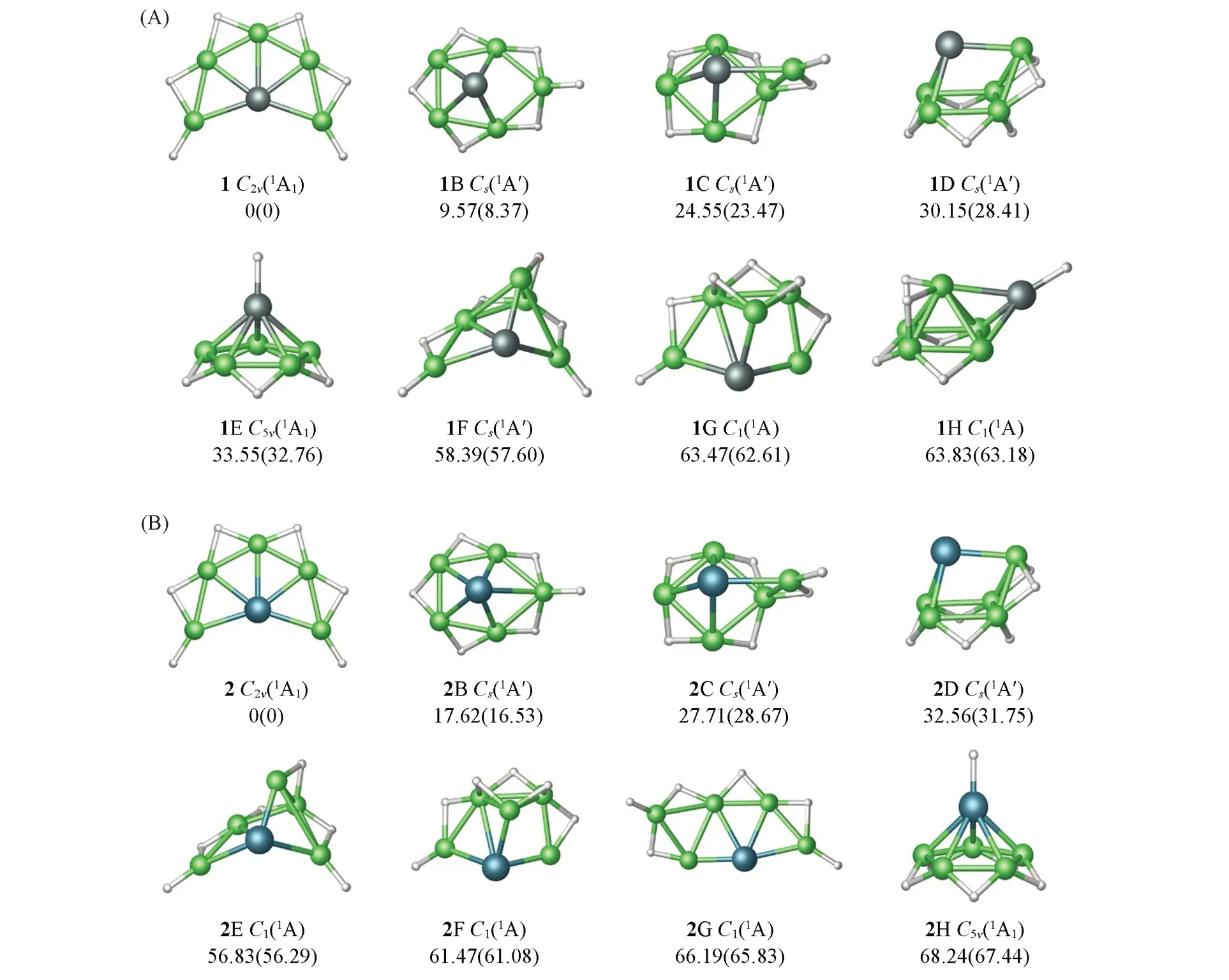
Fig.1 Optimized global minimum(GM)structures of SiBe5H 6(1)and GeBe5H 6(2)at the PBE0/aug⁃cc⁃pVTZ level along with seven lowest⁃lying isomer(n B—n H)
图2给出了PBE0/aug-cc-pVTZ理论水平上XBe5H6(X=Si,Ge)团簇全局极小结构的键长、韦伯键级及自然电荷.结构1和2中Si/Ge—Be键长介于0.213~0.226 nm,Be—Be键长介于0.192~0.207 nm,Be—H键长介于0.134~0.154 nm.依据Pyykkö[56]推荐的原子共价半径,Si/Ge—Be单键的上限为0.218/0.223 nm.需要注意的是,由于电负性差异,Si/Ge—Be成键有一定的极化.韦伯键级WBIX—Be=0.61~0.90,表明X与Be原子之间作用很强,接近于共价单键.Be—Be之间的韦伯键级WBIBe—Be=0.27~0.42.为了区分氢原子,桥氢和端基氢分别用Hb和Ht表示.Be—Hb之间的韦伯键级WBIBe—Hb=0.34~0.57,表明它们之间接近于共价半键;而WBIBe—Ht=0.87~0.88,揭示它们之间形成共价单键.结构1,2中ppSi/Ge中心携带-0.40|e|/-0.39|e|的负电荷;Be原子携带0.16~0.54的正电荷;而H原子携带-0.16~-0.33的负电荷.电负性较小的金属Be原子向电负性较高的H和Si/Ge有明显的电子转移,使得整体结构从内至外形成负-正-负三层结构.从静电作用角度来看,彼此吸引,有助于体系稳定.
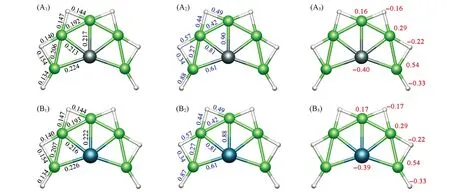
Fig.2 Bond distances(nm,A 1,B1),Wiberg bond indices(WBIs,A 2,B2),and natural atomic charges(|e|,A 3,B3)of SiBe5H 6(1)(A 1—A 3)and GeBe5H 6(2)(B1—B3)clusters at the PBE0/aug⁃cc⁃p VTZ level
平面五配位硅、锗团簇1,2为体系势能面上的全局极小结构,为了探讨其动力学稳定性,在PBE0/def2-SVP理论水平上对结构1和2进行了50 ps的BOMD模拟.图S1(见本文支持信息)给出了室温298 K条件下,XBe5H6(X=Si,Ge)(1,2)动力学模拟过程中相对于优化结构的均方根偏差(RMSD)曲线.由图S1可见,SiBe5H6的RMSD最高峰出现在28~31 ps处,主要是Si原子相对于Be5上下移动形成,表明Be5单元具有较大柔性.这一点与图2中的Be—Be键级(0.27~0.42)所得结论一致.已报道的准ptC-CBe4Au4,ppC-CBe4Li4团簇BOMD模拟中也有类似的情形,它们的RMSD平均值分别为0.032和0.043 nm[57,58].XBe5H6(X=Si,Ge)(1,2)整个BOMD模拟中,没有结构的突变,RMSD平均值分别为0.046和0.039 nm,表明它们动力学稳定性较好,没有异构化或解离.
2.2 化学键分析
为了进一步探讨平面XBe5H6(X=Si,Ge)团簇的稳定性与结构之间的关联,对代表性结构1(SiBe5H6)的正则分子轨道进行了分析.如图3所示,团簇SiBe5H6(1)含20个价电子,占据10个分子轨道.根据表S2(见本文支持信息)所列的原子轨道成分,可将其分子轨道分为4组.外围Be和H之间的作用源于图3(A)和(B)中两组轨道,而Be和中心Si原子的作用则源于图3(C)和(D)两组轨道.由图3(A)包括2个CMO(HOMO-3及HOMO-4),它们主要成分为Be的2s原子轨道和H原子的1s轨道.它们可通过组合形成2个Be—H二中心二电子(2c-2e)σ单键.图3(B)中有4个CMO(HOMO-5~HOMO-8).它们主要为Be(2s/2p)及桥H(1s)原子的贡献,可组合得到4个Be—H—Be 3c-2e键.图3(C)中仅有1个分子轨道,其为典型的离域π轨道,主要为Be2pz和Si3pz轨道的贡献.图3(D)中包括3个离域的σ轨道(HOMO-1,HOMO-2及HOMO-9),主要为Si3s/Si3p和Be2s/Be2p原子轨道的贡献.如此,SiBe5单元上有1个离域π轨道和3个离域σ轨道,它们提供了额外的稳定化能,有利于整体平面结构的稳定.GeBe5H6(2)的分子轨道图与之类似,如图S2及表S3(本文支持信息)所示.此外,在PBE0/aug-cc-pVTZ水平上结构1,2最高占据轨道(HOMO)和最低空轨道(LUMO)之间的能隙(HOMO-LUMO)均为3.93 eV,较大的能隙进一步支持了体系的稳定性.
AdNDP为传统NBO方法的新拓展,不仅可以和NBO一样分析定域轨道(1c-2e,2c-2e),而且还弥补了NBO方法无法处理三中心以上离域键的不足,能搜索出所有nc-2e键(n≤体系总原子数),从而更本质地揭示体系的成键特征.图4给出了PBE0/aug-cc-pVTZ水平上SiBe5H6(1)的AdNDP成键模式.图4(A)为SiBe5H6(1)中的端H与Be之间形成的2个二中心二电子(2c-2e)Be—Hσ键,电子占据数(ON)为1.99|e|;图4(B)为桥H与相邻Be原子之间形成的4个三中心二电子(3c-2e)Be—H—Be离域σ键,电子占据数均为1.99|e|.Be—H—Be离域σ键的存在有利于体系维持平面结构,进而有利于稳定平面五配位硅、锗.图4(C)中仅有一个覆盖整个SiBe5单元的6c-2e离域π键,电子占据数为2.00|e|.图4(D)为含3个6c-2e离域σ键,电子占据数为1.99~1.96|e|.这里需要指出的是,6σ电子本质是离域的,无法转化为定域的二中心二电子(2c-2e)路易斯键.对于平面团簇而言,电子离域有助于整个体系的稳定性.图4(C)和(D)中的4个离域键对于体系的稳定性至关重要:使SiBe5H6(1)团簇中心Si原子满足八隅律,即具有稳定的八电子壳结构;不仅于此,依据休克尔4n+2规则,离域的两个π电子使SiBe5单元具有π芳香性,而离域的6个σ电子则赋予其σ芳香性,即SiBe5H6(1)团簇具有2π和6σ双重芳香性.Boldyrev和Simons[59]在研究ptC CSi2Al2,CSi2Ga2及CGe2Al2系列五原子团簇时就指出,1个离域π键和3个离域σ键对于18电子的ptC体系稳定性十分重要.此外,18电子代表体系总电子数,成键包括外围配体原子之间的键和配体-ptC之间的键,其中最为重要的是配体-ptC之间的8个电子,简称“八电子”规则.八电子规则不仅适用于ptC团簇,还可拓展至ppC体系,如ptC CA及其衍生物;ppC及其衍生物.需要指出的是,这8个电子不必完全离域,部分定域也可以.最近,Cui等[33]通过电子定域的策略设计了系列新颖的ppSi团簇.这里SiBe5H6(1)团簇整体具有20价电子,稳定ppSi的主要为1个离域π键和3个离域σ键.这样看来,八电子规则不仅适用于平面四配位碳体系,对于平面五配位硅、锗体系同样有效.GeBe5H6(2)团簇的成键与SiBe5H6(1)团簇十分类似,如图S3(见本文支持信息)所示.
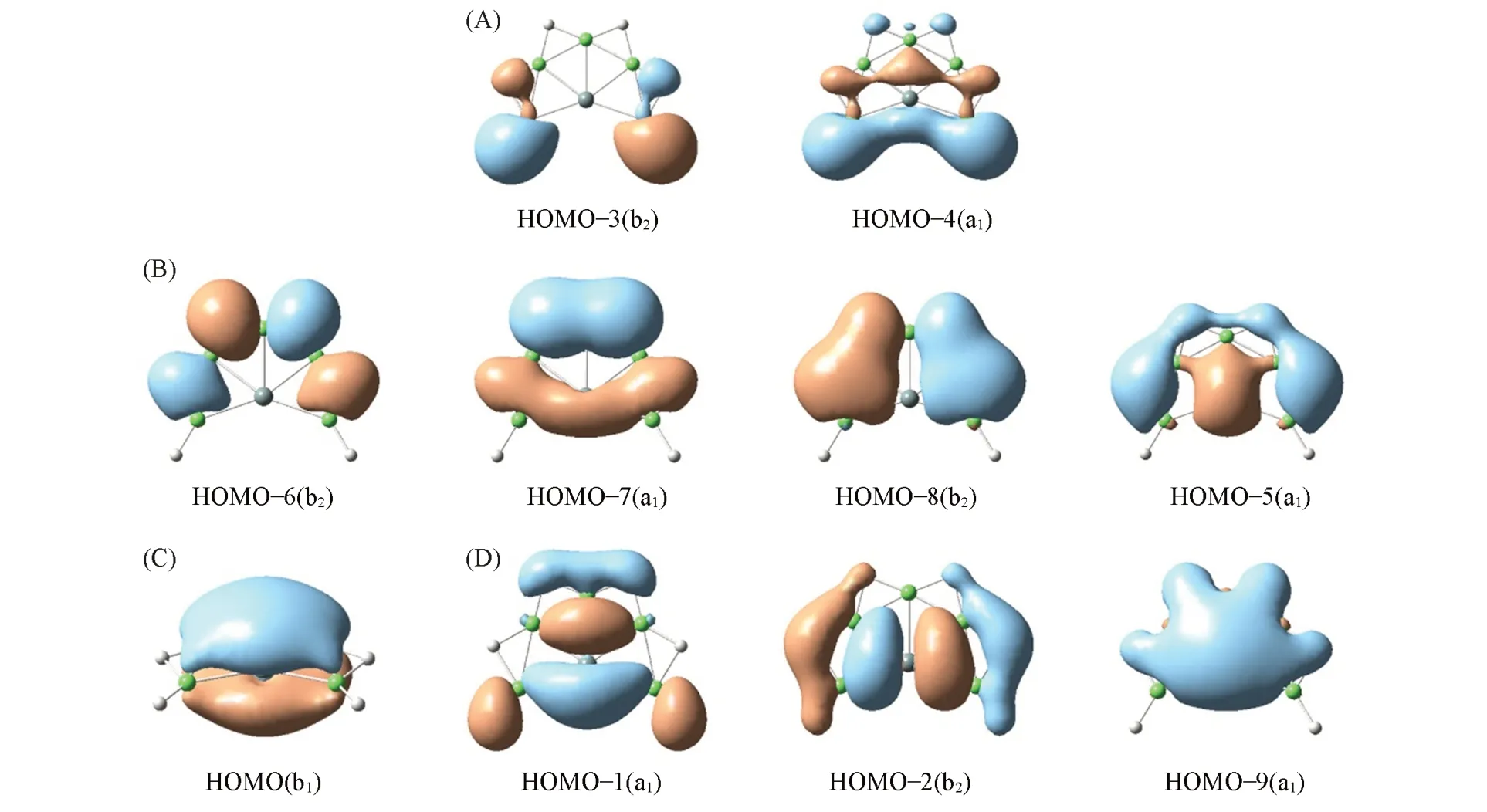
Fig.3 Canonical molecular orbitals(CMO)pictures of SiBe5H 6(1)
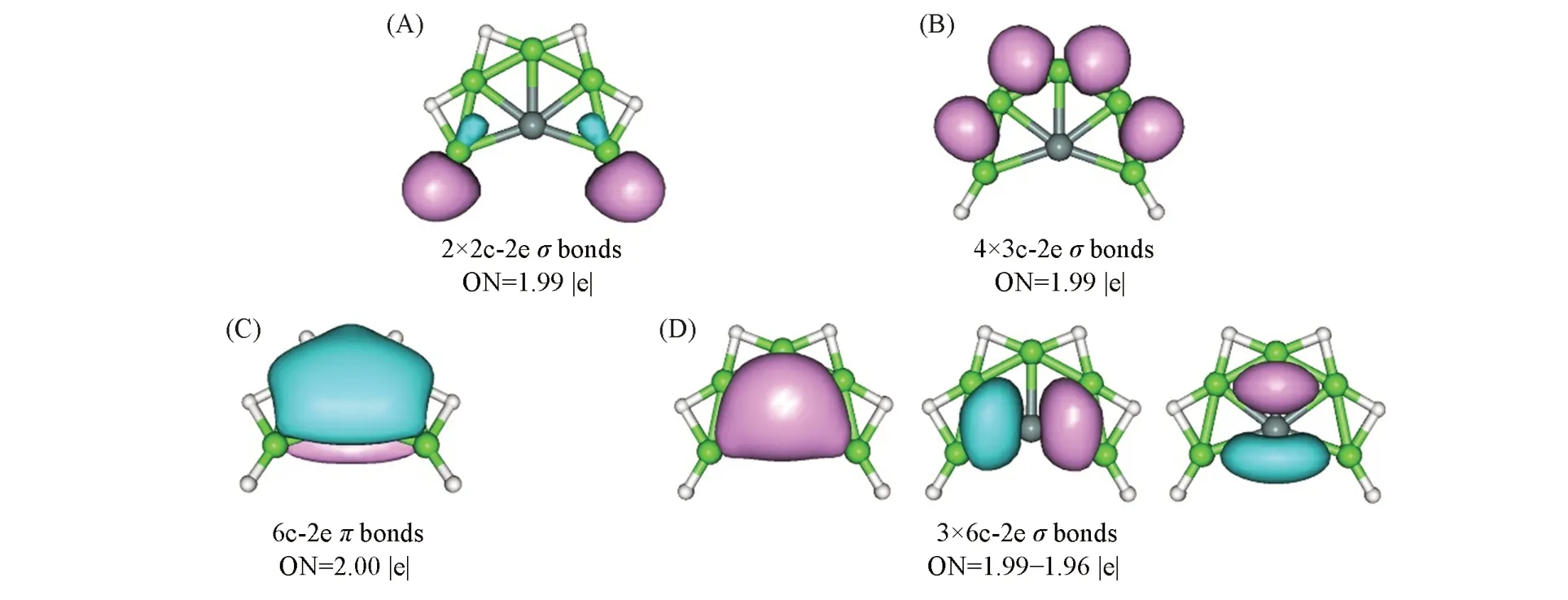
Fig.4 Adaptive natural density partitioning(Ad NDP)bonding scheme of SiBe5H 6(1)
为了定量化地揭示团簇中心原子与金属铍之间的相互作用,对SiBe5H6(1)和GeBe5H6(2)团簇进行了能量分解-化学价自然轨道(EDA-NOCV)分析.作用能(ΔEint)包括泡里排斥能(ΔEPauli)、静电作用能(ΔEelstat)及轨道作用能.以SiBe5H6(1)为例进行分析,将SiBe5H6分成Si和Be5H6两个碎片,并对各碎片可能的电荷及电子态进行全面考虑.如表S4(见本文支持信息)所示,对于SiBe5H6的EDA分析结果,当中性五重激发态的碎片和Be5H(6五重态)碎片相互作用时,SiBe5H6的轨道作用能(ΔEorb)最小.NBO分析揭示,SiBe5H6中Si的价电子分布为,进一步支持上述碎片

Table 1 EDA-NOCV results of XBe5H 6(X=Si,Ge)cluster at the PBE0/TZ2P//PBE0/aug-cc-p VTZ level,using X(3s13px13py13pz1)and Be5H 6(Quintet)as interacting fragments
图5给出了α,β电子及其复合的变形密度图.可见,α和β电子自旋轨道的电子流向刚好相反,表明Si和配体之间的作用本质为电子-共享键,其中仅存在少量的净电荷转移.第一组对应SiBe5H6的离域π轨道,Si和Be5H6配体之间基本为电子-分享键作用;第二、三组为Si3py,Si3px轨道与配体Be5H6的电子-分享键作用,其中Si反馈部分电子给Be5H6配体;第四组为Si3s轨道与Be5H6的电子-分享键作用,其中配体Be5H6提供部分电子给Si原子.如果将第二、三、四组变形密度加和,则可得到Si原子和配体σ型轨道作用的总变形密度.图5中的ΔEorb(2+3+4)更为清晰,5个Be原子和Si之间的电子在键区聚集,σ键作用本质为电子-共享键.图S4(见本文支持信息)给出了GeBe5H6的团簇形变密度(Δρ)图,与SiBe5H6的情形类似,仅有数据上的微小差别.此外,图S5(见本文支持信息)给出了代表性团簇SiBe5H6的能级相关图,五重激发态的Si原子通过4个单占轨道与Be5H(6五重态)的相应轨道进行组合,最终形成稳定的闭壳层体系SiBe5H6.
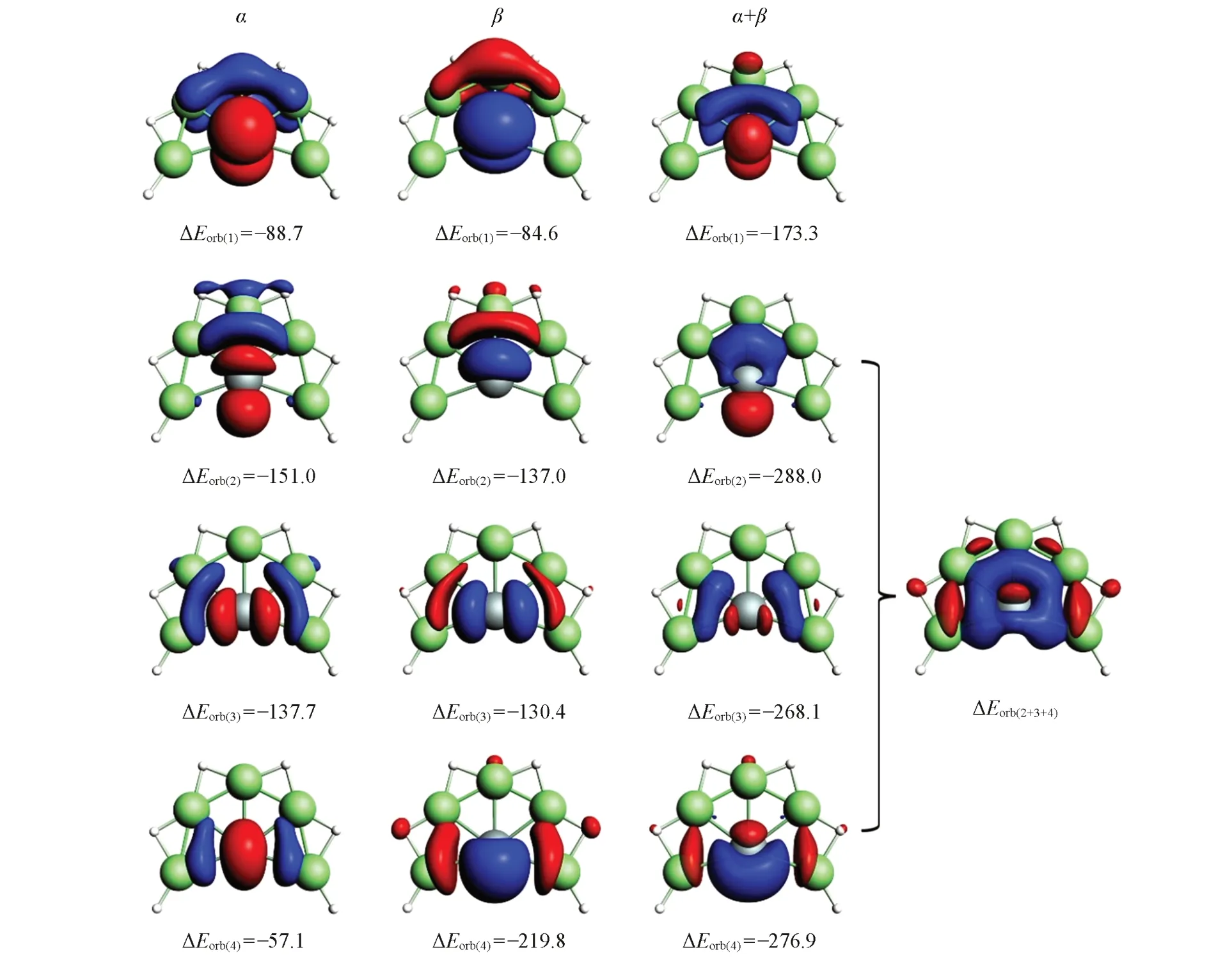
Fig.5 Plot of the deformation densities(Δρ)for EDA⁃NOCV analysis of SiBe5H 6(1)
2.3 双重芳香性
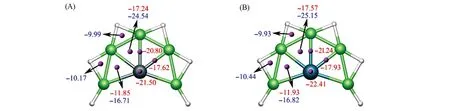
Fig.6 Nucleus independent chemical shifts(NICSs)for SiBe5H 6(1)(A)and GeBe5H 6(2)(B)clusters
为了进一步验证XBe5H6(X=Si,Ge)平面团簇的芳香性特征,对结构1和2的Be—H—Be,Be—X—Be三元环中心及其上方0.1 nm处,X原子、Be—X键上方0.1 nm处的核独立化学位移(NICS)值进行了计算.图6给出了PBE0/aug-cc-pVTZ理论水平上团簇1和2相应位置的NICS(0)及NICS(0.1)值,分别用蓝色和红色字体标注.NICS值可作为芳香性的半定量判据.NICS(0)和NICS(0.1)值分别用于评估体系的σ和π芳香性.按照AdNDP及分子轨道分析,团簇1和2外围Be—H—Be三元环仅具有σ芳香性,只列出NICS(0)数据;SiBe5,GeBe5单元所在的区域同时具有σ和π芳香性,因此NICS(0)及NICS(0.1)数据都列出,一些受原子核或化学键影响的NICS(0)不进行计算(如X原子及Be—X键中心处).由图6可见,NICS(0)及NICS(0.1)值均为较大的负值,表明团簇1和2具有σ和π双重芳香性.值得一提的是,最近报道的平面六配位Ga团簇GaBe6A也具有2π及6σ双重芳香性[60].如图S6(A)(见本文支持信息)所示,同种水平上,经典π芳香性苯分子环中心上方0.1 nm处的NICS(0.1)值为-9.98,而XBe5H6(X=Si,Ge)团簇的NICS(0.1)值(-11.85~-22.41)更负,进一步支持团簇1和2的芳香性结论.
与几个选定位置的NICS值相比,等化学屏蔽表面(Iso-chemical shielding surfaces,ICSS)可给出更为丰富、直观的信息.与NICS值不同的是,正的ICSS值表明体系具有芳香性.垂直于环平面的等化学屏蔽表面z方向分量(ICSSzz)比起ICSS在展现芳香性方面更有优势.图7为XBe5H6(X=Si,Ge)团簇平面ICSSzz及其上方0.1 nm处的ICSSzz磁屏蔽填色图.作为对照,图S6(B)给出了苯分子高于平面0.1 nm的ICSSzz填色图.由图7可知,XBe5单元具有π和σ双重芳香性,与之前CMO,AdNDP分析所得结论一致.
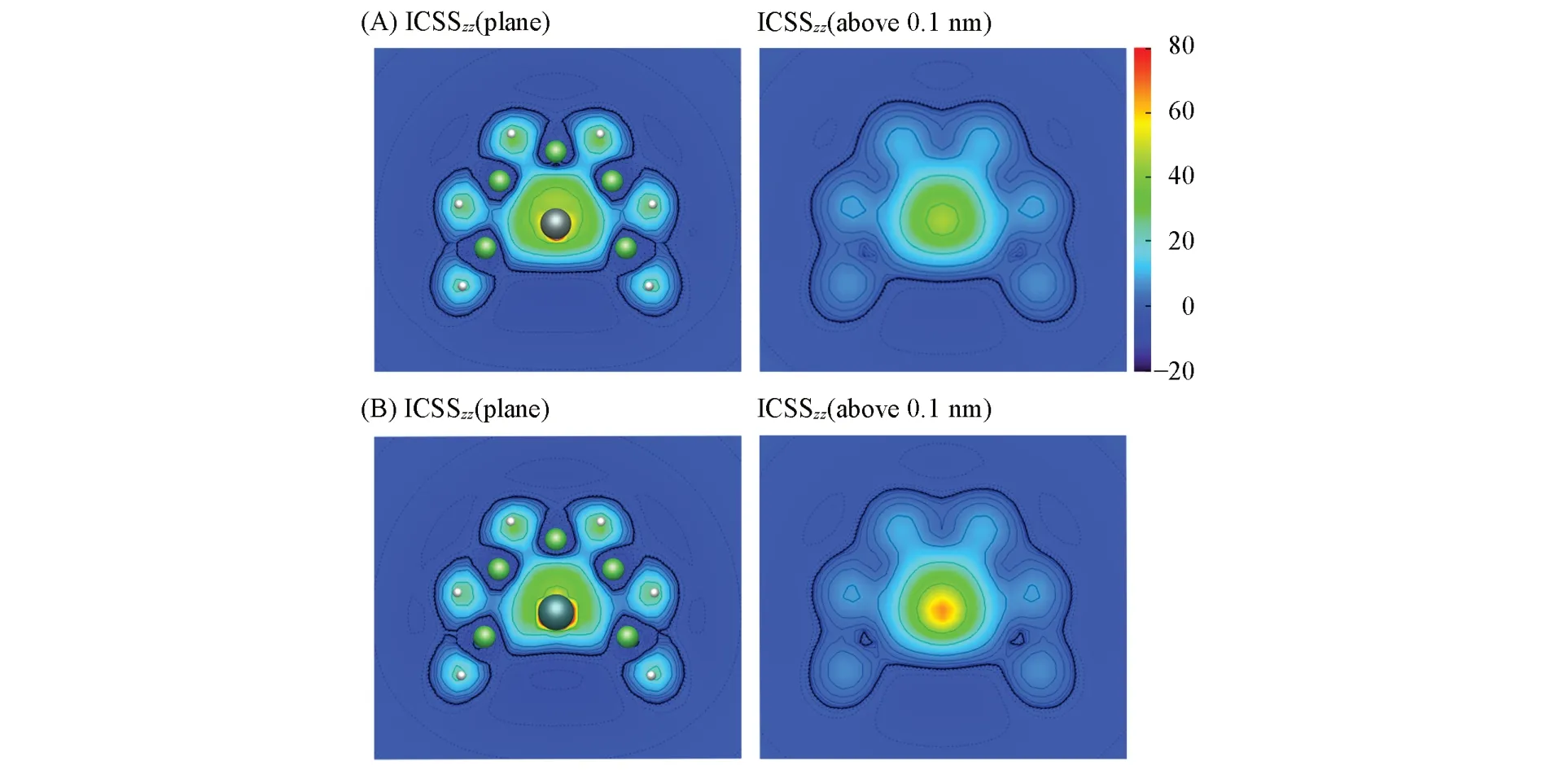
Fig.7 Color⁃filled maps of iso⁃chemical shielding surfaces in z direction,ICSS(0)zz and ICSS(0.1)zz for SiBe5H 6(1)(A)and GeBe5H 6(2)(B)
2.4 预测的红外光谱
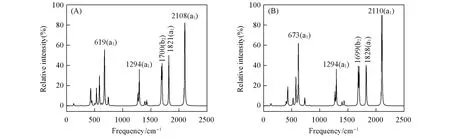
Fig.8 Simulated infrared spectra of SiBe5H 6(A)and GeBe5H 6(B)at the PBE0 level
为了便于进一步实验表征,在PBE0/aug-cc-pVTZ理论水平上对平面五配位硅、锗XBe5H6(X=Si,Ge)团簇的红外光谱进行了模拟.如图8所示,SiBe5H6(1)团簇光谱中最强吸收峰出现在2108 cm-1处,对应2个端基H和Be原子之间的伸缩振动.619 cm-1处的吸收峰为次强峰,主要源于Si—Be伸缩振动.1821和1700 cm-1处的吸收峰主要由桥H和Be原子之间的伸缩振动引起.其余的吸收峰相对较弱,多为复合振动峰.GeBe5H6(2)团簇的红外光谱特征与SiBe5H6(1)团簇基本类似,但由于中心原子不同,吸收峰的峰位、丰度略有差异.
3 结 论
理论设计了含平面五配位硅、锗的XBe5H6(X=Si,Ge)团簇,全局极小搜索及高精度量化计算结果表明,它们为全局极小结构.化学键分析揭示,XBe5核具有1个离域π键和3个离域σ键,外围4个Be—H—Be 3c-2e键及2个Be—H 2c-2e键.XBe5H6(X=Si,Ge)团簇具有特殊的2π/6σ双重芳香性.本文理论预测的平面五配位硅、锗团簇将进一步丰富平面多配位硅、锗的研究领域.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210807.
