抑制GRK2异常转膜控制JAK1-STAT1信号转导减少CIA小鼠树突状细胞的成熟
2022-06-08张春梅刘潇一杨雪枝
张春梅,斯 梦,刘潇一,杨雪枝,姜 玲,魏 伟
(安徽医科大学临床药理研究所、抗炎免疫药物教育部重点实验室、抗炎免疫药物安徽协同创新中心、安徽医科大学类风湿关节炎研究中心,安徽 合肥 230032)
类风湿关节炎(rheumatoid arthritis,RA)是一种慢性自身免疫性疾病,以关节对称性发展的慢性滑膜炎为特征,关节的持续炎症最终导致关节畸形甚至残疾[1]。RA的发病机制复杂,其中不同免疫细胞复杂的相互作用和活化参与RA疾病的发生发展。树突状细胞(dendritic cells,DCs)是体内功能最强大的抗原呈递细胞,可启动次级淋巴组织中的幼稚T细胞产生炎症因子,并浸润滑膜组织和滑液,在局部吸收、加工和呈递抗原,从而促进RA的持续存在[2]。IFN-α主要由浆细胞样DCs产生,研究发现,在一部分RA患者中Ⅰ型干扰素上调,IFN-α可以与IFNAR1和IFNAR2结合引起Janus激酶1(Janus kinase 1,JAK1)和tyrosine 激酶2(tyrosine kinase 2,TYK2)的激活,从而引起下游信号转导子和转录激活子1(signal transducers and activators of transcription,STAT1)/STAT2的磷酸化,随后p-STAT1/p-STAT2进入细胞核以启动炎症因子转录[3]。JAK-STAT信号通路已被确认参与RA中各种免疫细胞的激活和炎症细胞因子的释放。JAK抑制剂通过阻断JAK-STAT信号通路,从而抑制炎症因子的释放和免疫细胞的激活,在RA的治疗中取得了较好的治疗效果,但药物不良反应很多[4],这可能与炎症免疫反应的过度调节有关。因此,我们提出炎症免疫反应软调节,通过在不同细胞中寻找共同或相似的信号转导和网络调控的关键分子和特征,同时尽量地减少对细胞的伤害,选择性的将异常变化的细胞活性恢复到生理水平[5-6]。
G蛋白偶联受体(G protein-coupled receptors,GPCRs)是被广泛研究的药物靶点,主要是因为它们与人体生理、病理和药理活性密切相关。G蛋白偶联受体激酶2(G protein-coupled receptor kinase 2,GRK2)通过磷酸化活性受体的细胞内结构域来调节多种GPCR,导致受体脱敏和内化[7]。越来越多的数据表明,GRK2在RA等炎症性疾病中高表达,并参与调节炎症免疫细胞反应[8]。在生理条件下,GRK2主要位于细胞质中,炎症情况下GRK2向细胞膜转移,导致巨噬细胞极化[9]、成纤维样滑膜细胞异常增殖[10]、内皮细胞增殖迁移并促进血管生成[11],促进RA的发生发展。研究表明GRK2参与多种信号通路的调控,如PI3K/Akt、TLR、p38 MAPK、MEK1/2、ERK1/2等信号通路[6,12]。课题组研究发现,经IFN-α刺激后颌下腺细胞中GRK2与JAK1之间共定位减少[13],提示GRK2与JAK1之间可能存在联系,但是有关GRK2与JAK1-STAT1信号通路的关系尚未报道。本文主要通过建立小鼠胶原性关节炎(collagen-induced arthritis,CIA)模型和体外诱导DCs细胞模型研究GRK2与JAK1之间关系,为发现RA的新的病理机制和治疗靶点提供实验依据。
1 材料与方法
1.1 材料
1.1.1动物 SPF级DBA/1 ♂小鼠,7~8 w,(18±2)g,购自上海斯莱克实验动物有限责任公司,许可证号:SCXK(沪)2017-0005,饲养于安徽医科大学临床药理学研究所SPF动物实验室,饲养环境温度(20~25)℃,湿度(40%~70%),光照12 h。♂ BALB/c小鼠,6~8 w,购自安徽医科大学实验动物中心,许可证号:SCXK(皖)2017-001。
1.1.2试剂 鸡Ⅱ型胶原蛋白(CⅡ)(批号20011)购自美国Chondrex公司。RPMI 1640培养基(批号01-100-1ACS)和胎牛血清(批号04-001-1ACS)购自以色列BI公司。GSK180736A(批号HY-18990)和tofacitinib(批号477600-75-2)购自美国MedChemExpress公司。GRK2(批号sc-13143)和protein A/G琼脂糖(批号sc-2003)购自美国Santa公司。重组鼠干扰素-α(IFN-α)(批号752802)、APC-CD11c(批号117309)、FITC-MHCⅡ(批号107615)均购自美国BioLegend公司。流式抗体PE-CD83(批号130-104-474)、PE-Cy7-CD40(批号130-116-112)、PB450-CD86(批号130-123-279)均购自德国Miltenyi公司。PGE2(批号3632464)、重组鼠白介素4(rmIL-4)(批号021749)、重组鼠粒细胞巨噬细胞集落刺激因子(rmGM-CSF)(批号091855)均购自美国Peprotech公司。JAK1(批号ab133666)、p-STAT1(批号ab109461)抗体购自英国Abcam公司。p-JAK1(批号AF2012)、STAT1(批号AF6300)抗体购自美国Affinity公司,ELISA试剂盒PGE2(批号ml037542)、IFN-α(批号ml002017)、IL-6(批号ml002293-J)、TNF-α(批号ml002095-J)均购自上海酶联公司。
1.1.3仪器 Infinite M1000 PRO多功能酶标仪(瑞士TECAN公司);Image Quant化学发光成像系统(美国GE公司);Leica TCS SP8激光共聚焦显微镜(Leica公司);Leica DM4B 正置荧光显微镜(Leica公司);Beckman CytoFLEX流式细胞仪(Beckman 公司);ImageStreamX MarkⅡ成像流式细胞仪(Meck Millipore公司)。
1.2 方法
1.2.1小鼠CIA模型的建立 在无菌条件下,将鸡CⅡ溶解在乙酸(0.1 mol·L-1)中,终浓度为2 g·L-1,4 ℃过夜。BCG疫苗80 ℃灭活1 h,溶于液体石蜡,冰上研磨2 h,制备终浓度为2 g·L-1的弗氏完全佐剂(CFA)。二者等体积混合研磨乳化制成Ⅱ型胶原乳剂。于小鼠尾根部多处皮内注射0.1 mL,d 21后加强注射,d 29开始评分。
1.2.2小鼠整体指标评分 根据小鼠CIA模型评分标准进行评分,建立的关节炎指数评分标准如下,0分:无肿胀和局部发红迹象;1分:踝关节出现红斑及轻微肿胀;2分:踝关节至跖关节出现轻微红肿;3分:踝关节至跖关节出现中度红肿;4分:踝关节至跖关节出现中度红肿。对所有爪子进行评分,每只小鼠的最大可能关节炎指数评分为16。每只爪子都有五个指骨关节和一个踝关节或腕关节,因此每只小鼠的最大肿胀关节数为24。小鼠踝关节用石蜡包埋,切片进行HE染色,观察滑膜增生、淋巴细胞浸润、软骨破坏、血管翳生成的情况。
1.2.3流式细胞术检测CIA小鼠外周血和脾脏中DCs表型 使用淋巴细胞分离液分离出小鼠外周血和脾脏中的淋巴细胞,置于15 mL离心管中,加适量PBS清洗2遍,1 mL PBS重悬,血球计数板计数,取5×106个细胞,分别加入流式抗体CD11c、MHCⅡ、CD40、CD83,避光孵育30 min,加适量PBS离心洗去抗体,300 μL PBS重悬细胞,过滤网上机检测。
1.2.4免疫组化法检测小鼠脾脏中p-JAK1、p-STAT1、GRK2表达 将小鼠脾脏包埋切片,(1)脱蜡:将切片于60 ℃烘箱放置2 h,然后置于二甲苯浸泡2次(每次20 min)。(2)水化:100%乙醇浸泡2次(每次5 min),95%乙醇2 min,85%乙醇2 min,70% 乙醇2 min,双蒸水5 min,PBS洗3遍。加0.5% triton 通透20 min,PBS洗3遍。(3)抗原修复:EDTA修复10 min,PBS洗3遍,3% H2O220 min,PBS洗3遍。然后加3% BSA封闭1 h,PBS洗3遍,加p-JAK1(1 ∶50)、p-STAT1(1 ∶100)、GRK2(1 ∶50)一抗孵育过夜。PBS洗3遍,加二抗孵育20 min,PBS洗3遍,加DBA染色,苏木精复染,脱水,中性树脂封片。
1.2.5小鼠骨髓源DCs的培养 BALB/c小鼠处死后,于无菌环境中取出小鼠股骨,75%乙醇浸泡1 min,PBS冲洗2遍,用RPMI 1640培基冲出骨髓,6孔板铺板。于细胞培养箱中培养24 h后,留贴壁细胞,加入含GM-CSF(20 μg·L-1)和IL-4(20 μg·L-1)的RPMI 1640培基,隔天半量换液,补充GM-CSF和IL-4,d 7收集骨髓源DCs。
1.2.6ELISA检测CIA小鼠血浆中IFN-α、PGE2、IL-6、TNF-α以及DCs培养上清IL-6、TNF-α水平 收集小鼠外周血血浆和各组DCs培养上清,根据ELISA试剂盒使用标准检测IFN-α、PGE2、IL-6、TNF-α水平。
1.2.7流式细胞术检测DCs表型以及吞噬功能 收集d 7的DCs,于6孔板中培养,分为4组,加入IFN-α(40 μg·L-1)和PGE2(1 μmol·L-1)刺激24 h后,分别加入GRK2抑制剂(GSK180736A)(2 μmol·L-1)和tofacitinib(10 μmol·L-1)培养48 h。收集细胞于EP管中,2 000 r·min-1离心10 min,收集细胞上清留作ELISA样本。BMDCs中加入CD11c、CD40、CD83、CD86和MHCⅡ抗体,避光孵育30 min,加入PBS离心洗去抗体,300 μL的PBS重悬,过滤网后上机检测。吞噬功能检测步骤与表型检测一致,各组细胞等量分成两管,分别加入FITC-dextran(1 g·L-1)抗体100 μL,于4 ℃和37 ℃两种环境下,避光孵育90 min,加入PBS洗去抗体,300 μL的PBS重悬细胞,过滤网上机检测。
1.2.8Western blot法检测BMDCs中JAK1、p-JAK1、STAT1、p-STAT1以及细胞膜GRK2表达 DCs培养48 h后,收集DCs于EP管中,加入适量的细胞裂解液,提取总蛋白。总蛋白于100 000g下离心1 h,留沉淀,加入适量细胞裂解液溶解沉淀,得细胞膜蛋白。加入蛋白上样缓冲液后于100 ℃煮10 min,使蛋白变性。10%的SDS-PAGE凝胶电泳转膜,加JAK1、p-JAK1、STAT1、p-STAT1、GRK2抗体4 ℃孵育过夜,TPBS洗去一抗,二抗37 ℃孵育2 h,化学发光成像系统成像,ImageJ软件于分析灰度值。
1.2.9成像流式法检测BMDCs中p-STAT1的入核情况 同“1.2.7”收集DCs后,加入100μL的细胞固定液避光孵育10 min,PBS洗去固定液,之后加100 μL细胞破膜液,避光孵育15 min。除空白管外,其余各组加p-STAT1抗体室温孵育40 min,PBS洗1遍,弃去上清后,加荧光二抗避光孵育40 min,PBS洗去荧光二抗,加DAPI避光孵育5 min,随后加入PBS洗去DAPI,弃去上清,加200 μL的PBS重悬,过滤网上机检测。
1.2.10免疫共沉淀检测DCs中JAK1与GRK2共表达情况 同“1.2.8”收集细胞蛋白留用,取80 μL protein A/G珠置于EP管中,再加16 μL IgG抗体与protein A/G珠孵育2 h后,加COIP洗液清洗3遍,等量分为4管,分别加入各组细胞蛋白液孵育2 h,同时取80 μL protein A/G珠置于EP管中,再加16 μL GRK2抗体与protein A/G珠孵育2 h,取GRK2管,加COIP洗液清洗3遍,等量分为4管。取IgG管,3 000 r·min-1离心5 min,留上清加到GRK2管孵育16 h以上。3 000 r·min-1离心5 min,弃上清,留珠子,加COIP洗液清洗3遍,加入细胞裂解液和蛋白上样缓冲液,100 ℃煮10 min。后续步骤同“1.2.8”。

2 结果
2.1 CIA小鼠整体指标与炎症因子的变化小鼠CIA模型成功建立,并根据评分标准对关节炎指数、关节肿胀数进行评分,发现CIA小鼠的关节炎指数以及关节肿胀数在d 38~41达到高峰,然后关节肿胀开始消退。与正常组相比,CIA模型组小鼠关节炎指数与足爪肿胀度明显升高(P<0.01)(Fig 1A),对小鼠踝关节HE染色评分,CIA模型组有明显滑膜细胞增殖、淋巴细胞浸润、软骨侵蚀、血管翳生成(P<0.01)(Fig 1B)。ELISA结果显示,CIA模型组小鼠血浆中IFN-α、PGE2、IL-6、TNF-α含量明显上升(P<0.01)(Fig 1C)。

Fig 1 Overall indicators of CIA mice
2.2 CIA小鼠外周血和脾脏中DCs活化研究表明,RA患者体内DCs活化,并参与RA的发生发展。流式细胞术检测CIA小鼠外周血中DCs共刺激分子CD40、CD83的变化。流式细胞术结果表明,与正常组相比,CIA小鼠外周血中DCs表面共刺激分子CD40、CD83明显上调(P<0.01)(Fig 2A),CIA小鼠脾脏中DCs表面共刺激分子CD40(P<0.05)、CD83(P<0.01)同样上调(Fig 2B)。提示CIA模型小鼠的DCs处于活化状态。
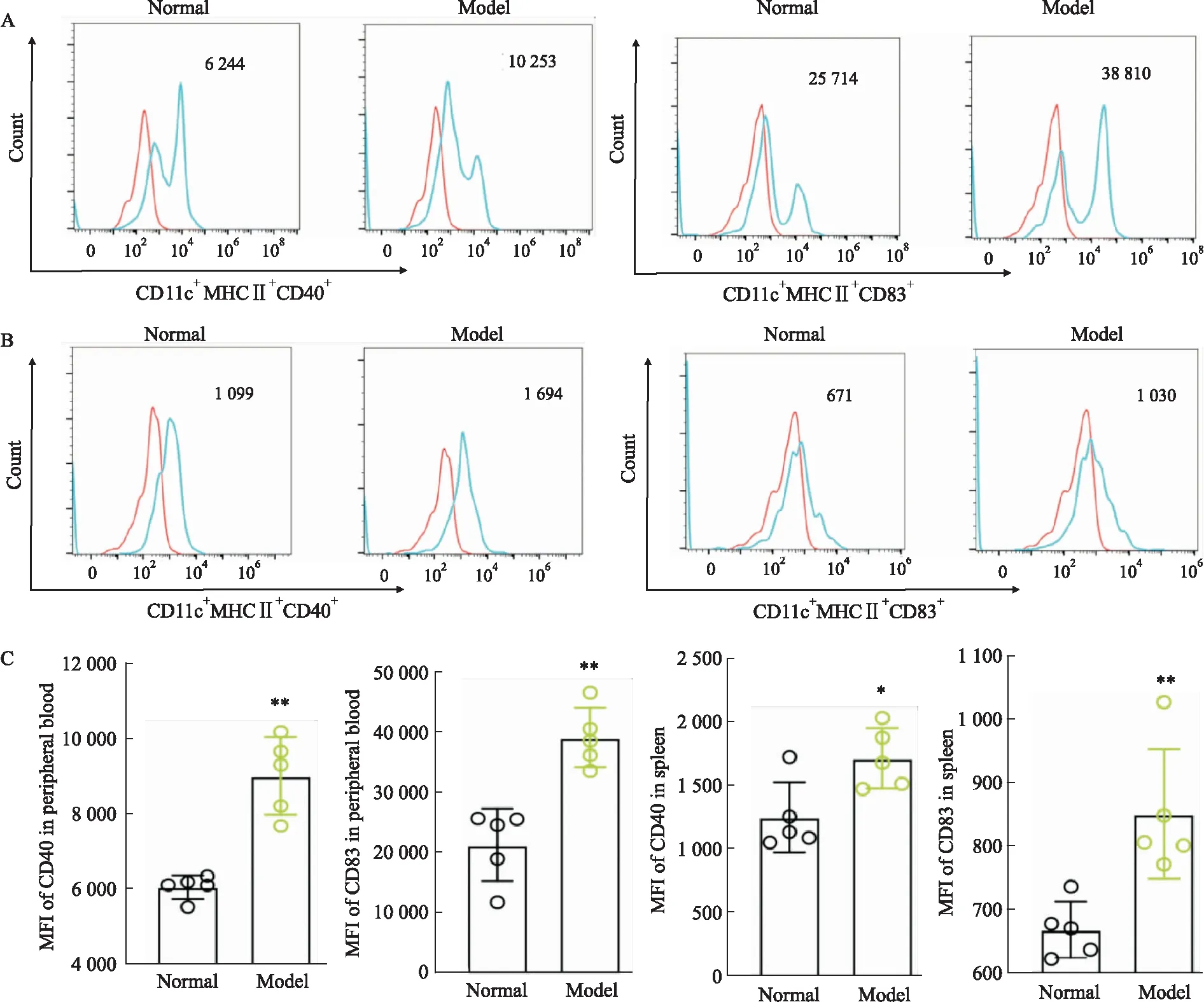
Fig 2 Expression of costimulatory molecules on surface of DCs detected by flow cytometry in CIA mice
2.3 CIA小鼠脾脏中p-JAK1、p-STAT1和GRK2的表达增加免疫组化法结果显示,CIA模型小鼠脾脏中p-JAK1、p-STAT1表达明显增加(P<0.01)(Fig 3A、3B)。课题组前期发现AA大鼠淋巴细胞中GRK2的表达增加[14]。免疫组化法检测小鼠脾脏GRK2表达的变化,CIA模型小鼠脾脏中GRK2表达明显增加(P<0.01)(Fig 3C)。相关性分析结果表明GRK2与p-JAK1、p-STAT1的表达呈正相关(Fig 4A)。为了进一步确定GRK2是否与JAK1-STAT1信号通路有关,免疫共沉淀结果表明,CIA模型组小鼠脾脏淋巴细胞中GRK2与JAK1的共表达明显下调(P<0.01)(Fig 4B)。

Fig 3 Expression of p-JAK1/p-STAT1 and GRK2 in spleen of CIA mice detected by

Fig 4 Correlation between GRK2 and JAK1-STAT1 signaling pathway
2.4 GRK2抑制剂调节DCs的功能为了确定GRK2和JAK1在DCs中的作用,我们将小鼠骨髓细胞诱导成DCs,使用IFN-α和PGE2刺激48 h模拟炎症环境,再给予GSK180736和tofacitinib培养24 h。为确认各组细胞活力是否有差异,使用CCK-8法对各组DCs的活力进行检测,结果显示,各组细胞活力无明显变化(Fig 5A)。ELISA和流式细胞术结果显示,与control组相比,刺激组DCs中炎症因子IL-6、TNF-α的水平升高(P<0.01),吞噬功能下调(P<0.01),其表面共刺激分子CD40、CD86、CD83、MHCⅡ水平上调(P<0.01),与IFN-α+PGE2组相比,GSK180736A下调DCs中炎症因子IL-6、TNF-α水平(P<0.01)(Fig 5B),上调DCs吞噬功能(P<0.01)(Fig 5C),并下调DCs中CD40(P<0.01)、CD86(P<0.01)、CD83(P<0.05)、MHCⅡ(P<0.05)的水平(Fig 6)。提示DCs的功能与GRK2的活性有关。

Fig 5 Effect of GRK2 inhibitor on secretion of inflammatory factors and phagocytosis of DCs
2.5 GRK2抑制剂对DCs中胞膜GRK2的表达以及GRK2与JAK1共定位的影响为了进一步确定GRK2对DCs中JAK1-STAT1信号通路的影响,Western blot结果表明,与control组相比,IFN-α+PGE2组DCs胞膜GRK2上调(P<0.01),GSK180736A下调DCs胞膜GRK2的表达(P<0.01)(Fig 7A)。免疫共沉淀结果表明,与control组相比,IFN-α+PGE2组GRK2与JAK1共表达减少(P<0.05),与IFN-α+PGE2组相比,GSK180736A组GRK2与JAK1共表达增加(P<0.05),而tofacitinib组并无变化(Fig 7B)。提示GRK2抑制剂通过抑制炎症环境下GRK2向细胞膜上的转移,将其留在胞质中,增加与JAK1的共定位。

Fig 6 Effect of GRK2 inhibitor on expression of CD40,CD83,CD86,and MHCⅡ on DCs treated with IFN-α and PGE2

Fig 7 Effect of GRK2 inhibitor on expression of GRK2 membrane in DCs and co-expression between GRK2 and JAK1
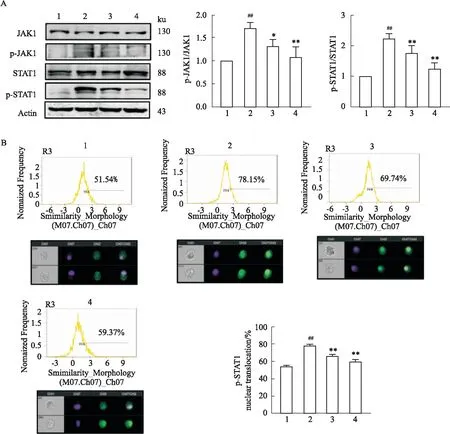
Fig 8 Effect of GRK2 inhibitor on JAK1-STAT1 pathway in DCs
2.6 GRK2抑制剂对DCs中JAK1-STAT1信号通路的影响为了进一步评估GRK2对DCs中JAK1-STAT1信号通路的调节作用,Western blot结果显示,与control组相比,IFN-α+PGE2组p-JAK1、p-STAT1表达增加(P<0.01),GSK180736A组p-JAK1(P<0.05)、p-STAT1(P<0.01)表达降低,tofacitinib组p-JAK1、p-STAT1表达降低(P<0.01)(Fig 8A)。成像流式细胞术结果显示,与control组相比,IFN-α+PGE2组p-STAT1入核率增加(P<0.01),与IFN-α+PGE2组相比,GSK180736A组和tofacitinib组p-STAT1入核率下调(P<0.01)(Fig 8B)。提示DCs功能的变化与GRK2调控JAK1-STAT1信号通路有关。
3 讨论
DCs作为体内功能最强大的抗原呈递细胞,参与RA的发病机制。在稳态情况下,DCs表现未成熟表型,其特征在于低表达共刺激分子和炎性细胞因子,当遇到外来抗原以及炎症环境刺激时,未成熟DCs分化成为成熟的DCs,高表达共刺激分子,并分泌大量炎症因子促进T细胞活化,成熟DCs浸润滑膜和关节组织,加重炎症免疫反应,从而促进RA的持续发展[15]。研究表明JAK-STAT信号通路在RA患者中激活。JAK抑制剂在RA的治疗中表现出较好的疗效,但长期使用会带来严重的不良反应,包括带状疱疹发病率增加、严重感染、静脉血栓栓塞、自然杀伤细胞数量减少、血小板减少和贫血等[4]。JAK抑制剂引起的不良反应可能与长期使用导致JAK-STAT信号功能丧失有关,JAK-STAT信号通路涉及机体各种生理功能,如JAK1信号缺失可引起神经系统疾病和严重淋巴细胞损伤、JAK2信号缺失可引起造血功能受损并导致胚胎死亡,JAK3的缺失引起严重的联合免疫缺陷、TYK2的缺陷可产生严重的过敏表型[4,16]。因此,需要恢复炎症信号通路正常转导,尽量减少或避免因过度抑制炎症信号通路而引发的不良反应。GRK2广泛存在于各种免疫细胞和非免疫细胞,主要位于细胞质,并调控多种信号通路的活化,如细胞质的GRK2可抑制内皮细胞中ERK1/2信号通路的激活,炎症情况下的GRK2向细胞膜转移导致ERK1/2信号通路活化,内皮细胞增殖和迁移能力增加,抑制GRK2异常转膜可恢复ERK1/2正常信号转导,从而降低内皮细胞的增殖和迁移能力[11]。但GRK2的转膜是否调控JAK1-STAT1信号转导尚不清楚,本研究主要探讨GRK2对DCs中JAK1-STAT1信号通路的调控作用。
我们构建了CIA小鼠模型,结果显示CIA模型小鼠外周血和脾脏中DCs共刺激分子CD40和CD83的表达上调,血浆中IL-6、TNF-α、PGE2和IFN-α的含量增加,这些结果表明DCs在CIA小鼠中被激活。而CIA小鼠脾脏中的JAK1-STAT1信号通路被激活,GRK2的表达增加,并且GRK2与JAK1的共定位在CIA小鼠脾脏中明显下调。提示GRK2与JAK1-STAT1信号通路存在相关性。
为了进一步确定GRK2与DCs中JAK1-STAT1信号通路的关系,我们在体外用骨髓细胞诱导DCs进行研究。结果表明,GRK2抑制剂降低了经IFN-α和PGE2的刺激的DCs中CD40、CD83、CD86和MHCⅡ的表达,增强抗原摄取能力,减少炎症因子分泌。另外有研究表明沉默STAT1抑制了CD40、CD83和CD86的上调,并减弱了TNF-α、IL-6和IL-12的分泌,这表明STAT1与DCs的功能有关[17]。实验结果显示,DCs经IFN-α和PGE2刺激后,GRK2在细胞膜表达增多,且GRK2与JAK1共定位减少,JAK1-STAT1信号通路激活,p-STAT1入核率增加,GRK2抑制剂抑制了GRK2向细胞膜的转移将其留在细胞质中,并增加了GRK2与JAK1的共定位,p-JAK1、p-STAT1表达减少,p-STAT1的入核率降低。这些结果表明,在IFN-α和PGE2的刺激下,GRK2发生异常转膜降低了对JAK1-STAT1信号通路的控制,通过抑制GRK2向细胞膜的转移,恢复JAK1-STAT1信号通路正常转导,从而减少DCs的成熟。
基于以上研究,在炎症条件下,细胞膜上的GRK2增加,JAK1-STAT1失去控制,引起下游信号异常转导。此外,课题组前期研究表明,EP4-cAMP信号通路被PGE2激活,GRK2转移至细胞膜引起EP4受体脱敏,导致cAMP水平降低,DCs成熟度增加[10,18]。当抑制GRK2向细胞膜转移时,EP4受体恢复敏感性,下游cAMP水平被恢复,同时留在胞质中的GRK2抑制JAK1的激活,从而降低DCs的成熟度。在本研究中,我们对GRK2调节DCs功能的作用有了更深入的了解,这可能与控制JAK1-STAT1信号转导有关。本文也为调控细胞异常活化恢复至生理水平的药物研究提供了新的依据。但GRK2调控JAK1的具体作用分子机制仍需要进一步探索。
