High-quality chromosome-level genome assembly of Tibetan fox (Vulpes ferrilata)
2022-06-07Tian-ShuLyu,Qin-GuoWei,Li-DongWang等
DEAR EDITOR,
Using Oxford Nanopore and Hi-C sequencing technology, we successfully assembled a chromosome-level genome of the Tibetan fox (Vulpesferrilata), with a total size of 2.38 Gb and N50 length of 133 960 477 bp.The 157 contigs were further assembled into 18 chromosomes with a sequence length of 2 378.42 Mb, accounting for 99.95% of the total length.A total of 21 715 protein-coding genes were predicted in the assembled genome, 86.47% of which were functionally annotated.Phylogenetic analysis showed thatV.ferrilataand the red fox (V.vulpes) formed a clade, with an estimated divergence time of 3.27 million years ago (Ma).Significantly enriched pathways and Gene Ontology terms associated with the expanded gene families in theV.ferrilatagenome were mainly related to hypoxia response and energy metabolism,indicating the mechanistic strategy ofV.ferrilatafor highaltitude adaptation.Furthermore, selection signature analysis identified genes associated with DNA damage repair and angiogenesis inV.ferrilata.Construction of theV.ferrilatagenome provides valuable information for further genetic analysis of important biological processes, which will facilitate the study of genetic changes during evolution.
The Tibetan fox, which belongs to the family Canidae and order Carnivora, is widely distributed in the northern Qinghai-Xizang (Tibet) Plateau (QTP) at altitudes of more than 3 500 m a.s.l.(Clark et al., 2008).As a plateau-endemic species, it plays an important role in maintaining ecological balance(Harris et al., 2014).Since the 1990s, concerted conservation efforts have led to the rapid increase inV.ferrilatapopulations, which have played a vital role in preventing and controlling local rodent pests (Liu, 2013).Despite this, various questions remain to be clarified.For example, the divergence time betweenV.ferrilataand the red fox (V.vulpes) remains controversial (ranging from 2.43 to 4.74 Ma) (Fritz et al., 2009;Humphreys & Barraclough, 2014) and whetherV.ferrilatahas evolved a unique plateau adaptation mechanism that differs from other species is unclear.The assembly of high-quality genomes should help clarify the adaptation mechanisms of high-altitude animals and provide a useful resource for future research.Thus, we constructed a high-quality genome assembly ofV.ferrilataby combining next-generation sequencing (NGS) short-read, Nanopore long-read, and Hi-C read sequencing.We compared the genomic features ofV.ferrilatawith those of 10 other species, focusing on hypoxia response- and energy metabolism-related gene families,which may contribute to high-altitude adaptation and resistance to hypoxic and low-temperature conditions.
We collected an adult maleV.ferrilatasample for sequencing from Gande County, Golog Tibetan Autonomous Prefecture, Qinghai Province (N33°54'1", E99°48'54").Permits for sample collection and use are provided in the Supplementary Materials.Leg muscle tissue was collected for DNA extraction and heart, liver, lung, gut, testis, and kidney tissues were collected for total RNA extraction.In this research, different strategies were used to obtain different omics data.For long-read genome sequencing (PromethION,library size: 20 kb), we obtained ~253.01 Gb of Nanopore reads (read number: 15 437 356; mean read length: ~16.39 kb; N50: ~23 kb) after base calling using Guppy v3.2.2 (-c dna_r9.4.1_450bps_fast.cfg).The MGISEQ2000 platform(library size: 400 bp) was used to obtain the NGS data.After quality control using fastp 0.20.0 (-n 0 -f 5 -F 5 -t 5 -T 5), 145 Gb of NGS short-insert reads were obtained (Supplementary Tables S1, S2).We also obtained ~264 Gb of Hi-C reads from the Illumina NovaSeq 6 000 platform (pair-ended sequencing with a read length of 150 bases and library size of 350 bp).For transcriptome data, we obtained ~46.03 Gb of clean data(Supplementary Table S3).Additional details are provided in the Supplementary Materials and Methods.The 17-mer frequency distribution analysis was performed using Jellyfish v2.2.10, which revealed that the estimated genome size ofV.ferrilatawas ~2.3 Gb (Supplementary Figure S1).We alsoobtained a preliminary ~2.35 Gb genome assembly (contig N50: 61 Mb) using NextDenovo v2.0-beta.1 (reads_cutoff:1k,seed_cutoff:25k).Data correction was performed using Racon v1.3.1 and NextPolish v1.0.5, and the final polished genome assembly was ~2.38 Gb with a contig N50 of ~61.59 Mb(Supplementary Table S4).Genome completeness and accuracy were evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO) v4.0.5 (-l mammalia_odb10 -g genome), with 92.76% complete BUSCOs (8 558) identified(Supplementary Table S5).GC depth analysis was used to assess the presence of exogenous contamination in the genome.Results revealed a genome coverage depth of 102.73 X and average GC content of 41.25% (Supplementary Figure S2).The single nucleotide polymorphism (SNP) and indel tests identified 8 282 homozygous SNPs, accounting for 0.000 348% of the genome (depth>=5X).The number of homozygous indels was 29 587, accounting for 0.001 243% of the genome (depth>=5X).Thus, the genome showed 99.998 409% base accuracy (depth>=5X) (Supplementary Table S6).Overall, the assessment results indicated high accuracy and completeness of theV.ferrilatagenome assembly.
For chromosome-level assembly, HIC Pro v2.8.1 was used to control the quality of the raw Hi-C data.Due to interruption and error correction of contigs during Hi-C-assisted assembly,the number of contigs eventually changed from 359 to 379.Finally, 157 contigs (2 340 706 585 bp) were anchored onto 18 linkage groups with a mounting rate of 98.36%(Supplementary Table S7; Figure 1A).The final assembly results are presented in Supplementary Table S8.To better understand theV.ferrilatagenome assembly, basic genomic information was compared with that of otherVulpesspecies(Table 1).Of the three species, theV.ferrilatagenome had the largest contig N50 (Kukekova et al., 2018; Peng et al.,2021).Analysis showed that theV.ferrilataandCanislupus familiarisgenomes had a high degree of synteny (Figure 1B),consistent with their close phylogenetic relationship.
GMATA v2.2 and Tandem Repeats Finder v4.07b (2 7 7 80 10 50 500 -f -d -h -r) were used to identify tandem repeats(TRs).Transposable elements (TEs) were identified using MITE-Hunter (-n 20 -P 0.2 -c 3), RepeatModeler v1.0.11 (-engine wublast), and RepeatMasker v1.331 (nolow -no_is -gff-norna -engine abblast -lib lib).We identified a total of 4 545 261 repeats with a size of ~857.2 Mb using repeat annotation, accounting for 37.77% of the assembledV.ferrilatagenome (Supplementary Table S9 and Figure S3).
Three independent methods were used for gene prediction:i.e.,ab initioprediction, homology search, and transcriptome prediction.Specifically, for the prediction of RNA-seq-based genes, STAR v2.4 was used to align filtered mRNA-seq reads with the reference genome.The transcript assembled by Stringtie v1.3.4d and PASA v2.3.3 was used to predict open reading frames (ORFs).GeMoWa v1.6.1 was used to align homologous peptides of related species with the assembled transcript to obtain gene structure information for homologous prediction.Forde novoprediction, RNA-seq-based sequences were assembled using Stringtie v1.3.4d, and a training set was generated using PASA v2.3.3.Augustus v3.3.1 was then used forab initiogene prediction of the training set.EvidenceModeler v1.1.1 (EVM) was used to combine the three results, for a total of 21 715 protein-coding genes(Supplementary Table S10).
After aligning the obtained genes in four databases,including Non-Redundant Protein Sequence Database (NR),Gene Ontology Resource (GO), Clusters of Orthologous Groups for Eukaryotic Complete Genomes (KOG), and Kyoto Encyclopedia of Genes and Genomes (KEGG), BUSCO evaluation showed that the genome benchmark reached 86.47%, suggesting an almost complete assembly.In total,18 776 genes were functionally annotated (Supplementary Table S11 and Figure S4).We also annotated different types of non-coding RNAs (ncRNAs) based on the Rfam database.The prediction results indicated that the ncRNAs of theV.ferrilatagenome included 143 320 transfer RNAs (tRNAs),3 788 microRNAs (miRNAs), 295 ribosomal RNAs (rRNAs),and 585 regulatory RNAs (Supplementary Table S12).The annotated gene set ofV.ferrilatawas compared to that of other species.Analysis showed that the annotated genes of these species had similar distribution trends, indicating a reliable annotation of theV.ferrilatagenome (Supplementary Figure S5).
Gene families forV.ferrilataand other species (i.e.,Ailuropodamelanoleuca,Canislupus,Enhydralutris,Felis catus,Homosapiens,Leptonychotesweddel,Musmusculus,Mustelaputorius,Pantheratigris, andV.vulpes) were clustered using OrthoMCL v2.0.9 (Li et al., 2003).Finally,5 835 single-copy genes were obtained and used for phylogenetic inference (Supplementary Figure S6).Although the phylogenetic tree topology is consistent with previous phylogenetic studies, the divergence time betweenV.ferrilataandV.vulpesdiffered (3.27 Ma) (Fritz et al., 2009; Humphreys& Barraclough, 2014) (Supplementary Figure S7).This divergence time coincides with the geological events related to the rapid uplift of the QTP three Ma, indicating a more reliable divergence time estimation for theVulpesgenus (Zhong &Ding, 1996).
We compared the expansion and contraction of gene families in theV.ferrilatagenome with that of 10 other species(including eight species in the order Carnivora) using CAFÉ v4.2.1.Among the 419 expanded gene families inV.ferrilata,79 changed significantly (P<0.05), involving 538 genes(Figure 1C).After KEGG and GO enrichment analysis of the expanded orthologous groups, 67 significantly overrepresented pathways and 32 significantly enriched GO terms were obtained (Figure 1D, E; Supplementary Tables S13,S14).Notably, extreme temperature response- and hypoxiarelated gene families showed significant pathway expansion,including the oxidative phosphorylation (ko00190, 44 genes,P=1.32E-35), thermogenesis (ko04714, 17 genes,P=1.81E-05), mTOR signaling pathway (ko04150, 44 genes,P=2.02E-34), HIF-1 signaling pathway (ko04066, 21 genes,P=7.28E-10), vascular smooth muscle contraction (ko04270,12 genes,P=2.24E-05), and calcium signaling pathway(ko04020, 10 genes,P=0.005 373 021) families.The 32 significantly enriched GO terms, including ATP hydrolysiscoupled proton transport (GO:0015991, 44 genes,P=2.18E-53), oxidoreductase activity (GO:0016620, 33genes,P=1.17E- 23), glucose metabolic process(GO:0006006, nine genes,P=5.64E-13), NAD binding(GO:0051287, nine genes,P=5.84E-09), cellular iron ion homeostasis (GO:0006879, seven genes,P=4.92E-07), and protein ubiquitination (GO:0016567, three genes,P=0.025 884), were related to energy metabolism and transportation and may contribute to the adaptation ofV.ferrilatato low oxygen and extreme temperature conditions.Thus, the significantly expanded genes associated with hypoxia and energy metabolism may reflect mechanisms underlying the adaptations ofV.ferrilatato high-altitude environments.

Table 1 Comparison of genome assemblies between V. ferrilata and other Vulpes species
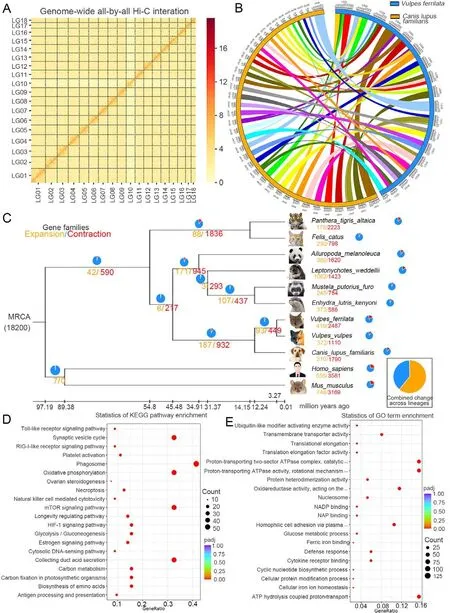
Figure 1 Statistics and data analysis of genome assembly of Vulpes ferrilata
The Codeml program in PAML v4.8 was employed to test for positively selected genes (PSGs) in theV.ferrilatagenome using the branch site model (Yang, 2007).We identified 175 PSGs inV.ferrilata(Supplementary Table S15).Of these PSGs, nine may be related to the adaptation ofV.ferrilatato low oxygen and high UV radiation, including vascular endothelial growth factor A (VEGFA) and mitochondrial genome maintenance exonuclease 1 (MGME1).More detailed information is provided in Supplementary Table S16.Compared with other native plateau species, the adaptation strategy ofV.ferrilatais reflected more significantly at the expanded gene family level than in PSGs.This suggests thatV.ferrilatamay have a unique plateau adaptation mechanism,which requires further exploration.
To the best of our knowledge, this is the first report on the chromosome-level genome ofV.ferrilata, a species unique to the QTP.We also determined the divergence time betweenV.ferrilataandV.vulpes.Furthermore, analysis of expanded and contracted gene families and PSGs revealed the possible adaptation strategies ofV.ferrilatato the plateau environment.This high-quality genome provides a solid basis for future studies on the population and conservation of this species and will help to improve our understanding of the environmental adaptations of species native to the QTP.
SCIENTIFIC FIELD SURVEY PERMISSION INFORMATION
Permission for field surveys in Qinghai Province was granted by the Qinghai Forestry and Grassland Bureau.Project approval of administrative license (2021: No.7) was issued by the Qinghai Forestry and Grassland Bureau.All sample collection procedures and experiments were approved by the Qinghai Forestry and Grassland Bureau and conformed to the guidelines established by the Ethics Committee for the Care and Use of Laboratory Animals of Qufu Normal University(Permit No.QFNU2018-013).
DATA AVAILABILITY
The genome assembly and sequenced data were submitted to the Science Data Bank databases (DOI 10.11922/sciencedb.01523), National Center for Biotechnology Information (NCBI: PRJNA762184, PRJNA768296, and JAJBZS000000000), and National Genomics Data Center(GSA: CRA006096; GW: GWHBHNE00000000).
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
T.S.L.conceived the project.T.S.L., T.G., and L.D.W.collected the samples.Q.G.W., S.Y.Z., Y.H.D., and L.P.S.performed the genome assembly, gene annotation, and bioinformatic analysis.T.S.L.wrote the manuscript.W.L.S.,H.S.D., and H.H.Z.revised the manuscript.All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We thank Mr.Ting-Bin Lyu for assistance with sample collection.We are particularly grateful to Jiangwen Cairen for assistance in this study.We would also like to thank the Qinghai Forestry and Grassland Bureau for support during this project.
杂志排行

Zoological Research的其它文章
- Adult hippocampal neurogenesis and its impairment in Alzheimer’s disease
- Discovery of a wild, genetically pure Chinese giant salamander creates new conservation opportunities
- Combinational benefit of antihistamines and remdesivir for reducing SARS-CoV-2 replication and alleviating inflammation-induced lung injury in mice
- Characterization of two novel knock-in mouse models of syndromic retinal ciliopathy carrying hypomorphic Sdccag8 mutations
- Scan of the endogenous retrovirus sequences across the swine genome and survey of their copy number variation and sequence diversity among various Chinese and Western pig breeds
- Novel astrovirus and paramyxovirus in Mongolian gerbils (Meriones unguiculatus) from China