Characterization of two novel knock-in mouse models of syndromic retinal ciliopathy carrying hypomorphic Sdccag8 mutations
2022-06-07ZhiLinRenHouBinZhangLinLiZhengLinYangLiJiang
Zhi-Lin Ren, Hou-Bin Zhang,3, Lin Li,3, Zheng-Lin Yang,3,*, Li Jiang,3,*
1 Department of Laboratory Medicine, Sichuan Provincial People's Hospital, School of Medicine, University of Electronic Science and
Technology of China, Chengdu, Sichuan 610072, China
2 Sichuan Provincial Key Laboratory for Human Disease Gene Study, Sichuan Provincial People's Hospital, University of Electronic Science and Technology of China, Chengdu, Sichuan 610072, China
3 Research Unit for Blindness Prevention of Chinese Academy of Medical Sciences (2019RU026), Sichuan Academy of Medical Sciences,Chengdu, Sichuan 610072, China
ABSTRACT Mutations in serologically defined colon cancer autoantigen protein 8 (SDCCAG8) were first identified in retinal ciliopathy families a decade ago with unknown function.To investigate the pathogenesis of SDCCAG8-associated retinal ciliopathies in vivo, we employed CRISPR/Cas9-mediated homology-directed recombination (HDR) to generate two knock-in mouse models,Sdccag8Y236X/Y236X and Sdccag8E451GfsX467/E451GfsX467,which carry truncating mutations of the mouse Sdccag8, corresponding to mutations that cause Bardet-Biedl syndrome (BBS) and Senior-Løken syndrome (SLS) (c.696T>G p.Y232X and c.1339-1340insG p.E447GfsX463) in humans,respectively.The two mutant Sdccag8 knock-in mice faithfully recapitulated human SDCCAG8-associated BBS phenotypes such as rod-cone dystrophy, cystic renal disorder, polydactyly, infertility, and growth retardation, with varied age of onset and severity depending on the hypomorphic strength of the Sdccag8 mutations.To the best of our knowledge,these knock-in mouse lines are the first BBS mouse models to present with the polydactyly phenotype.Major phototransduction protein mislocalization was also observed outside the outer segment after initiation of photoreceptor degeneration.Impaired cilia were observed in the mutant photoreceptors,renal epithelial cells, and mouse embryonic fibroblasts derived from the knock-in mouse embryos, suggesting that SDCCAG8 plays an essential role in ciliogenesis, and cilium defects are a primary driving force of SDCCAG8-associated retinal ciliopathies.
Keywords: SDCCAG8; Primary cilia; Retinal ciliopathy; Bardet-Biedl syndrome (BBS); Senior-Løken syndrome (SLS); Nephronophthisis (NPHP);Polydactyly
lNTRODUCTlON
Retinal ciliopathies are a group of inherited retinal degenerative diseases caused by mutations in genesencoding ciliary proteins essential for photoreceptor morphology and function (Adams et al., 2007; Bachmann-Gagescu & Neuhauss, 2019; Bujakowska et al., 2017; Chen et al., 2021).Retinal ciliopathies can present as non-syndromic retinal disorders such as Leber congenital amaurosis (LCA) or retinitis pigmentosa (RP) (Hartong et al., 2006; Koenekoop et al., 2007; Kumaran et al., 2017; Verbakel et al., 2018), as well as syndromic retinal dystrophies, such as Bardet-Biedl syndrome (BBS), Senior-Løken syndrome (SLS), Joubert syndrome (JBTS), Meckel-Gruber syndrome (MKS), Jeune syndrome, and Alström syndrome (AS) with the involvement of multiple systems and organs, including the central nervous system, kidney, skeleton, liver, and adipose tissue (Braun &Hildebrandt, 2017; Hurd & Hildebrandt, 2011; Zaghloul &Katsanis, 2009).Over 100 genes encoding retinal ciliopathy proteins are associated with retinal ciliopathies, accounting for almost 25% of all retinal dystrophies (Chen et al., 2019)(https://sph.uth.edu/retnet/).
Serologically defined colon cancer autoantigen protein 8(SDCCAG8) was identified as a causative gene of retinal ciliopathy following detection of its mutation in retinal-renal ciliopathy families a decade ago (Otto et al., 2010).Genotypephenotype correlation studies have suggested that SDCCAG8-associated syndromic ciliopathies manifest predominantly as retinal-renal degeneration, accompanied by obesity, hypogonadism, recurrent pulmonary infections,cognitive defects, and mild intellectual disability in some cases, but not with polydactyly (Halbritter et al., 2013b; Kang et al., 2016; Otto et al., 2010; Schaefer et al., 2011) (Table 1).Thus, theSDCCAG8gene is alternatively referred to asNPHP10,SLS7, andBBS16.Recent genome-wide association studies have also demonstrated that genetic polymorphisms of SDCCAG8 are associated with bipolar disorder and schizophrenia (Gonzalez et al., 2016; Hamshere et al., 2013).
The humanSDCCAG8gene encodes a 713 residue fulllength protein that contains a N-terminal globular domain(1-270 amino acids (aa)), short nuclear localization signal,and large C-terminal coiled-coil domain (CCD) (Kenedy et al.,2003; Otto et al., 2010).To date, 19 retinal ciliopathy-causingSDCCAG8mutations have been identified, including deletion,insertion, nonsense, and splicing mutations across exon5 to exon16 of the gene, resulting in a reading frame shift that produces C-terminal CCD-truncated proteins of different sizes(Table 1).Previous genotype-phenotype correlation studies have revealed thatSDCCAG8truncating mutations near the N-terminal are predominantly associated with BBS, while ones near the C-terminal are primarily associated to SLS (Table 1).However, it has been difficult to correlate specific truncating mutations to phenotypic variants in SDCCAG8-associated syndromic retinal ciliopathies, and the underlying pathogenesis remains largely unknown.

Table 1 SDCCAG8 mutations identified in patients with retinal ciliopathies
Vertebrate photoreceptors possess a highly specialized primary sensory cilium, composed of a basal body (BB),connecting cilium (CC)/transition zone (TZ), and outer segment (OS), where retinal ciliopathy proteins localize and function (May-Simera et al., 2017).SDCCAG8 localizes at the BB/centrosome and CC/TZ in photoreceptors, and in other ciliated cell types in other systems, and directly interacts with other ciliopathy-associated proteins, including OFD1, NPHP5,RP1, and RPGRIP1 (Di Gioia et al., 2012; Insolera et al.,2014; Otto et al., 2010; Patil et al., 2012).Suppression of SDCCAG8 expression in zebrafish causes developmental defects in the kidney, brain, and body axis (Otto et al., 2010).Sdccag8mutant mouse models carrying distinct gene-trap alleles, i.e.,Sdccag8gt,Sdccag8tm1e(EUCOMM)Wtsi, andSdccag8SBT, show loss of full-length SDCCAG8 protein expression (Airik et al., 2014; Insolera et al., 2014; Weihbrecht et al., 2018).Sdccag8gt/gtmice present with early-onset retinal degeneration, late-onset nephronophthisis (NPHP), as well as developmental and structural abnormalities of the skeleton and limbs, mimicking disease phenotypes in humans (Airik et al., 2016, 2014 ).In addition, theSdccag8tm1e(EUCOMM)Wtsi/tm1e(EUCOMM)WtsiandSdccag8SBT/SBTmouse lines exhibit neonatal lethality with developmental defects in the central nervous system, limbs, and lungs, but without retinal-renal involvement, a major feature of SDCCAG8-assciated ciliopathies (Insolera et al., 2014;Weihbrecht et al., 2018).Thus, these threeSdccag8mutant mouse models show significant phenotype variation and genotype-phenotype discrepancies.
To investigate how differentSDCCAG8truncating mutations may cause syndromic retinal ciliopathies with different severity and system involvement, we employed CRISPR/Cas9-mediated homology-directed recombination (HDR) technology to generate knock-in mouse modelsSdccag8Y236X/Y236XandSdccag8E451GfsX467/E451GfsX467carrying truncating mutations of the mouseSdccag8gene, which correspond to BBS- and SLS-causing mutations in humans, respectively.Results showed that the two mutantSdccag8knock-in mice closely phenocopied retinal and renal degeneration of SDCCAG8-associated retinal ciliopathies, with varying disease onset and severity.In addition, the mice displayed preaxial polydactyly,which is absent in retinal ciliopathies caused bySDCCAG8mutations.They also showed major phototransduction protein mislocalization outside the OS after initiation of photoreceptor death.Retinal photoreceptors, renal epithelial cells, and mouse embryonic fibroblasts (MEFs) from the knock-in mice exhibited impaired biogenesis and structural defects in cilia,suggesting that SDCCAG8 plays an essential role in ciliogenesis, and its dysfunction is an underlying mechanism driving retinal and renal degeneration inSdccag8knock-in mice.
MATERlALS AND METHODS
Mutant Sdccag8 knock-in mice
All procedures for animal experiments were approved by the Animal Care and Use Committee of Sichuan Provincial People’s Hospital and conformed to the recommendations of the Association for Research in Vision and Ophthalmology(Approval No.2014NSF(09)).Mice were maintained under 12 h cyclic dark/light conditions.
We generated two knock-in mouse models carrying either a point mutationSdccag8-Y236X (c.708C>G p.Y236X) in exon7 or a 1 bp insertionSdccag8-E451GfsX467 (c.1 351-1352insG p.E451GfsX467) in exon11 with CRISPR/Cas9-mediated HDR technology (Viewsolid Biotech, China).The correspondingSDCCAG8mutations in humans, i.e.,c.696T>G p.Y232X and c.1339-1340insG p.E447GfsX463,are known to cause BBS and SLS, respectively (Otto et al.,2010).We first designed two mutation-specific guide RNA(gRNA) targets, i.e.,Sdccag8-Y236X-g (5'-GGCTGAA ACTCACATACGAGG-3') andSdccag8-E451GfsX467-g (5'-ACGTTGCGTCTCAGGAAATGG-3'), to guide sequencespecific cutting near each mutation, as well as two corresponding donor DNA oligos, i.e.,Sdccag8-Y236X-d (5'-TCCTGCCTTGTTCTGCAGGAGAGGCTGAAACTCACATAG GATCCGGCGAAGACTGACCTTCTGGAATCTCAGCTGATG CTT-3') andSdccag8-E451GfsX467- d (5'-GTCACTTAGAGGAAATTCAGAACCCGTTGCGTCTCAAGG ATCCAATGGACGTCACAAAGGTCCGAGAAAGTTTTGCTTT AA-3') to introduce the mutations into mouseSdccag8genomic DNA through HDR.In addition, we designed a restriction endonuclease (BamHI) site (GGATCC) next to each mutation to facilitate mouse genotyping, as well as a synonymous mutation 1350G>A inSdccag8-E451GfsX467-d to destroy the protospacer adjacent motif sequence to prevent recurrence of gRNA-mediated cutting following recombination.Each gRNA and donor DNA oligo pair was microinjected into the fertilized eggs of C57BL/6 mice together with CRISPR plasmid encoding Cas9 nuclease to generateSdccag8knockin chimeras through CRISPR/Cas9-mediated HDR.Founders with successful recombination were selected by Sanger sequencing with mouse tail DNA, and subsequently used to produce heterozygous and homozygous knock-in miceSdccag8Y236X/Y236XandSdccag8E451GfsX467/E451GfsX467for our study.
Mouse genotyping
Knock-in allelesSdccag8-Y236X andSdccag8-E451GfsX467 were identified by polymerase chain reaction (PCR)amplification with subsequent BamHI restriction enzyme digestion due to the introduction of a BamHI site next to each mutation.Two pairs of primers were designed for allelespecific PCR: i.e.,Sdccag8-Y236X-F:ACAGCAGAGTGGAGTGAGCTAGT andSdccag8-Y236X-R:TTGAGCACAGGAGACACCTAAC;Sdccag8-E451GfsX467-F:GCTGAGAAGGTAGAGAAGTG andSdccag8-E451GfsX467-R: CACACCACACCACATACAT.PCR amplification was conducted in a total reaction volume of 20 μL.Subsequently,8.7 μL of PCR raw products were directly digested using the BamHI restriction enzyme (New England Biolabs (NEB), USA)in a 10 μL reaction system at 37 °C for 1 h, and then analyzed by 2% agarose gel electrophoresis.
DNA constructs and transfection
We obtained pEGFP-Sdccag8plasmids expressing the mouse wild-type SDCCAG8 protein with the N-terminal fluorescent protein marker enhanced green fluorescent protein (EGFP)(OriGene, USA).For the generation of pEGFP-Sdccag8(Y236X) and pEGFP-Sdccag8(E451GfsX467) mutant constructs, we designed two pairs of mutagenesis primers,pSdccag8(Y236X)-F: TCCGGCGAAGACTGACCTTCT and pSdccag8(Y236X)-R: TCCTATGTGAGTTTCAGCCTCTC;pSdccag8(E451GfsX467)-F: GATCCAATGGACGTCACAAA GGTG and pSdccag8(E451GfsX467)-R:CTTGGGACGCAACGTGGTTCTGA.We then conducted QuikChange XL site-directed mutagenesis with mutagenesisprimers following the manufacturer’s protocols (Stratagene,USA).The wild-type and mutant plasmids were transfected into HEK293T cells (cultured with 10% fetal bovine serum(FBS)/Dulbecco’s Modified Eagle Medium (DMEM) containing 1% penicillin-streptomycin solution, 37 °C, 5% CO2) with a Lipofectamine 3 000 Kit (Invitrogen, USA).Expression of SDCCAG8 wild-type and mutant proteins in the transfected cells was analyzed by western blotting.
Preparation of MEFs
The MEFs were derived from wild-type andSdccag8knock-in E13.5 embryos, as described previously (Dong et al., 2015),then cultured with 20% FBS/DMEM containing 2% penicillinstreptomycin at 37 °C in humidified 5% CO2.To analyze cilium biogenesis, the MEFs were seeded in 24-well plates and serum-starved for 24 h before immunofluorescence staining.
Histology
The eyeballs and kidneys were dissected from euthanized(CO2inhalation) mice and fixed in fixative solution of 1.22%glutaraldehyde and 0.8% paraformaldehyde in 0.08 mol/L phosphate buffer at 4 °C overnight.Subsequently, the fixed tissues were dehydrated through an ethanol series and embedded in paraffin.Sections were taken at 5 μm for both retinas and kidneys.Hematoxylin and eosin staining was performed for retinal and kidney sections, and Masson trichrome staining was conducted for kidney sections following standard protocols.Images of the stained sections were acquired with a Zeiss Axiovert 200 microscope (Carl Zeiss,USA) under 63× objective.Images across the entire retinal sections were acquired with a 20× objective and imported into ImageJ v1.8.0 software with 3.8 pixels/mm scaling.The outer nuclear layer (ONL) thicknesses were measured at 500 μm intervals from the optic nerve head.
Transmission electron microscopy (TEM)
Photoreceptor ultrastructure inSdccag8knock-in mice was investigated using standard protocols, as described previously(Jiang et al., 2011).Dissected mouse eyecups were first fixed in 2.5% glutaraldehyde and 1% paraformaldehyde in 0.1 mol/L cacodylate buffer overnight at 4 °C, then postfixed with 1%osmium tetroxide in 0.1 mol/L cacodylate for 1 h.The fixed eyecups were staineden blocwith uranyl acetate after bufferwashing and dehydrated with methanol solutions.The eyecups were subsequently embedded in Epon812 resin (Ted Pella, USA) and cut into 60 nm sections with an ultramicrotome (Leica EM UC7, Germany) near the optic nerve.The sections were placed onto carbon-coated copper grids and stained with both uranyl acetate and lead citrate for contrast enhancement.TEM was performed at 75 kV using a H-7 650 electron microscope (Hitachi-Science & Technology,Japan) to observe the morphology of the photoreceptor ciliary compartments.
Western blotting
Mouse retinas and transfected cells were lysed by sonication in standard RIPA buffer (150 mmol/L NaCl, 1% Triton X-100,0.5% sodium deoxycholate, 0.1% SDS, 50 mmol/L Tris-HCl pH 7.4) supplemented with complete protease inhibitor cocktail tablets (Roche, USA).The supernatant of protein lysates (~20 μg) was resolved using 10% sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to a nitrocellulose membrane (0.45 μmol/L,Millipore, Germany).Subsequently, the membrane was blocked with 8% non-fat dry milk in Tris-buffered saline with 0.1% Triton X-100 (TBST) for 2 h at room temperature and then probed with primary antibodies overnight at 4 °C,followed by horseradish peroxidase (HRP)-conjugated second antibodies at room temperature for 1 h.
lmmunochemistry
Mouse eyeballs were dissected from euthanized mice and immediately fixed with 4% paraformaldehyde (PFA) in 0.1 mol/L phosphate buffer (pH 7.4) for 2 h on ice, then dehydrated sequentially with 15% and 30% sucrose in 0.1 mol/L phosphate buffer (pH 7.4) for 2 h each.The eyecups were then embedded in optimal cutting temperature compound (OCT, Sakura Finetek, USA) after lens removal and cut into 12 μm retinal sections.Mouse kidneys were first dissected and fixed with 4% PFA.After dehydration with 30%sucrose for 24 h, the kidneys were embedded in OCT and cut into 5 μm sections.MEFs cultured on coverslips were fixed in 4% PFA for 10 min and permeabilized with 1% Triton X-100 for immunostaining.
For immunofluorescence staining, retinal and kidney sections, as well as MEF slides, were blocked with 10%normal donkey serum and 0.2% Triton X-100 in phosphate buffer at room temperature for 1 h, then incubated with primary antibodies overnight at 4 °C.After washing three times with phosphate buffer, the tissue sections and MEF slides were incubated with fluorescence-conjugated secondary antibodies at room temperature for 1 h.To investigate apoptosis of mouse retinas, TUNEL staining of retinal sections was performed with anIn SituCell Death Detection Kit (Roche Diagnostics, China).Fluorescence images were acquired using a confocal microscope (LSM800,Carl Zeiss, Germany).
Antibodies
The primary antibodies used for western blotting and immunofluorescence analysis included: SDCCAG8 (WB 1:2 000, IHC 1:50, 13 471-1-AP; Proteintech Group, USA),GRK1 (1:400, 24 606-1-AP; Proteintech Group, USA),rhodopsin (1:400, 1D4, clone D4B9B; Cell Signaling Technology, CST, USA), S-opsin (1:300, Ab5407; Abcam,UK), GFP (1:3 000, 50 430-2-AP; Proteintech Group, USA),GAPDH (1:5 000, 10 494-1-AP; Proteintech Group, USA), βactin (1:5 000, 20 536-1-AP; Proteintech Group, USA), PDE6B(1:400, T13343; Thermo, USA), Alexa Fluor 594 conjugated peanut agglutinin (PNA) (1:200, L32459; Thermo, USA), cone arrestin (1:300, AB15282; Sigma, USA), and anti-alpha tubulin(acetyl K40) (1:1 000, ab24610; Abcam, UK).Secondary antibodies included: goat anti-rabbit Alexa Fluor 488 and 594(1:1 000; Invitrogen, USA), goat anti-mouse Alexa Fluor 594(1:1 000; Invitrogen, USA), and HRP-conjugated Affinipure goat anti-rabbit IgG (H+L) (1:5 000, SA00001-2; Proteintech Group, USA).
Electroretinogram (ERG) analysis
As described previously (Jiang et al., 2011), experimentalmice were dark-adapted overnight, with subsequent procedures performed under dim red light.The dark-adapted mice were first anesthetized with a combination of ketamine(16 mg/kg body weight) and xylazine (80 mg/kg body weight)by intraperitoneal injection, and their eyes were then dilated with tropicamide, phenylephrine, and tetracaine (0.5%).Body temperature was maintained at 37 °C with a heating pad.Both scotopic and photopic ERG responses were recorded from 3-5 mice with the Espion Visual Electrophysiology System(Diagnosis, AbelConn, LLC, USA).Scotopic ERG,representing rod visual function, was first conducted with light stimuli at intensities ranging from -2.52 log cd·s/m2to 1.30 log cd·s/m2, while photopic ERG, representing cone visual function, was performed with light stimuli at intensities ranging from 0.48 log cd·s/m2to 1.30 log cd·s/m2after light adaptation for 20 mins.Antibiotic ointment was applied to the eyes after the ERG procedure to prevent infection.
Analysis of urine albumin to creatinine ratio (uACR)
Proteinuria, which is a major feature of chronic kidney disease(CKD), was assessed based on the uACR.We obtained 24 h urine samples from mice at P180 using metabolic cages (TSE systems, Germany).The concentrations of urine microalbumin and creatinine were measured by immunoturbidimetric assay(Mindray, China) and kinetic enzymatic assay (Maccura,China), respectively, on an AU5800 automatic biochemical analyzer (Beckman Coulter, Japan).uACR was calculated by dividing the microalbumin concentration in micrograms by the creatinine concentration in milligrams, reported as μg/mg.
Statistical analysis
T-test was used to compare data between two groups.Multiple comparisons involving more than three groups were analyzed using analysis of variance (ANOVA).Significance was determined atP<0.05.Data were analyzed using GraphPad Prism v8 (GraphPad Software, USA) and are presented as mean±standard error of the mean (SEM).
RESULT S
Generation of two knock-in mouse models expressing hypomorphic alleles, Sdccag8-Y236X and Sdccag8-E451GfsX467
Two humanSDCCAG8recessive mutations, i.e., nonsense mutation 696T>G (p.Y232X) and 1 bp insertion 1 339-1340insG (p.E447GfsX463), are known to cause syndromic retinal ciliopathies BBS and SLS, respectively (Table 1)(Otto et al., 2010).To investigate genotype-phenotype correlation and pathogenesis of SDCCAG8-associated retinal ciliopathiesin vivo, we utilized CRISPR/Cas9-mediated HDR to generate two knock-in mouse models carrying the corresponding mouse mutationsSdccag8-Y236X andSdccag8-E451GfsX467 (Figure 1A-C).
The knock-in mouse mutations were verified by Sanger sequencing (Figure 1D, E).To facilitate knock-in mouse genotyping, we designed a BamHI restriction site (GGATCC)adjacent to the mutations in the HDR donor oligos and performed BamHI digestion of allele-specific PCR amplificons as a routine genotyping procedure.Thus, there were two mutant allele-specific bands (258 bp and 170 bp) and one wild-type allele-specific band (425 bp) inSdccag8-Y236X mouse genotyping (Figure 1F).Similarly, there were two mutant allele-specific bands (263 bp and 186 bp) and one wild-type allele-specific band (445 bp) inSdccag8-E451GfsX467 mouse genotyping (Figure 1G).All three bands were present in the heterozygous mice.
Upon western blotting with anti-SDCCAG8 antibodies targeting the N-terminal epitope (1-360 aa) of SDCCAG8, we detected 27 kDa and 54 kDa truncated protein bands inSdccag8Y236X/Y236XandSdccag8E451GfsX467/E451GfsX467mouse retinas at P30, respectively, but the absence of the full-length SDCCAG8 protein at 83 kDa (Figure 2A).Molecular weights of the mutant proteins, SDCCAG8-Y236X and SDCCAG8-E451GfsX467, were consistent with the predicted truncations due to reading frame shift of theSdccag8mutations.The expression levels of both truncated proteins were significantly decreased in the knock-in mice compared to that of the fulllength protein in wild-type mice, indicating instability of the truncated proteins (Figure 2A).To confirm whether truncation and reduction of the SDCCAG8 mutant proteins could be attributed to the disease-causing mutations rather than other genetic engineering modifications via CRISPR/Cas9-mediated HDR, such as insertion of a BamHI restriction site, we expressed GFP-tagged SDCCAG8 wild-type and mutant proteins in HEK293T cells.Using immunoblotting analysis with anti-SDCCAG8 and anti-GFP antibodies, we confirmed the expression of truncated proteins in their corresponding transfected cells, consistent with theSdccag8mutants expressed inSdccag8knock-in mice at reduced levels(Figure 2B).To investigate the localization of truncated SDCCAG8 in mouse photoreceptors, we conducted immunochemical assays on mouse retinas at P30 (Figure 2C).SDCCAG8 was localized around the photoreceptor inner segment (IS) and CC in wild-type controls, as reported previously (Otto et al., 2010), whereas the truncated SDCCAG8 proteins were expressed in the same location in the mutant photoreceptors but with significantly deceased fluorescence signals (Figure 2C).Thus, we verified the two knock-in mouse lines carrying hypomorphicSdccag8mutant alleles,Sdccag8Y236XandSdccag8E451GfsX467.
The two knock-in mouse lines displayed unexpected Mendelian ratios, with only 9.1% ofSdccag8Y236X/Y236Xmice(40/433) and 17.4% ofSdccag8E451GfsX467/E451GfsX467mice(83/476) surviving after birth, significantly lower than the expected ratio of 25%.Additionally, 45% ofSdccag8Y236X/Y236Xmice (18/40) and 42.2% ofSdccag8E451GfsX467/E451GfsX467mice(35/83) died within 24 h of birth.TheSdccag8knock-in mice had normal body size at birth but were significantly smaller than the age-matched controls at P30, indicating developmental retardation (Figure 2D).Compared to the controls (17.67±1.37 g), theSdccag8E451GfsX467/E451GfsX467mice(10.50±1.38 g) and especially theSdccag8Y236X/Y236Xmice(9.33±1.21 g) had significantly reduced body weight(Figure 2E).Notably, all mutant males were infertile.
Development of early-onset and progressive rod-cone degeneration with varied severity in two Sdccag8 knockin mouse models
Previous studies have reported that individuals withSDCCAG8-associated ciliopathies manifest early-onset and progressive retinal degeneration (Otto et al., 2010).To examine whether theSdccag8knock-in mice phenocopied human retinal degeneration caused bySDCCAG8mutations,we performed histological analysis on mouse retinas at ages P30, P90, and P180 (Figure 3A-C).In contrast to wild-type controls with 11-12 rows of photoreceptor nuclei lining the retinal ONL, theSdccag8Y236X/Y236XandSdccag8E451GfsX467/E451GfsX467mice had only around six and eight rows, respectively, at P30 (Figure 3A, D).In addition,their photoreceptor OSs were about half as short as that of the wild-type controls at P30 (Figure 3A).Shortening of the ONL and OSs progressed rapidly in the knock-in mice from P30 to P180.TheSdccag8E451GfsX467/E451GfsX467mice had only around six rows of photoreceptor nuclei remaining in the ONL at P90,and close to four rows at P180 (Figure 3B-F).TheSdccag8Y236X/Y236Xmice showed more severe retinal degeneration, with only four rows of photoreceptor nuclei left at P90, and two rows at P180 (Figure 3B-F).Correspondingly,their photoreceptor OSs were substantially shortened by P90 and almost completely diminished at P180, indicating rapid retinal degeneration between P30 and P180 in the knock-in mice.To examine whether theSdccag8mutations also caused degeneration of cone photoreceptors, we used PNA staining, a cone specific marker, on knock-in mouse retinas at P30, P90, and P180 (Figure 3G).There was no obviousreduction in cone photoreceptors, even by P90, in both knockin mice, althoughSdccag8Y236X/Y236Xmice showed significant loss of cones and reduced cone OS at P180, revealing lateonset cone generation inSdccag8knock-in mice.
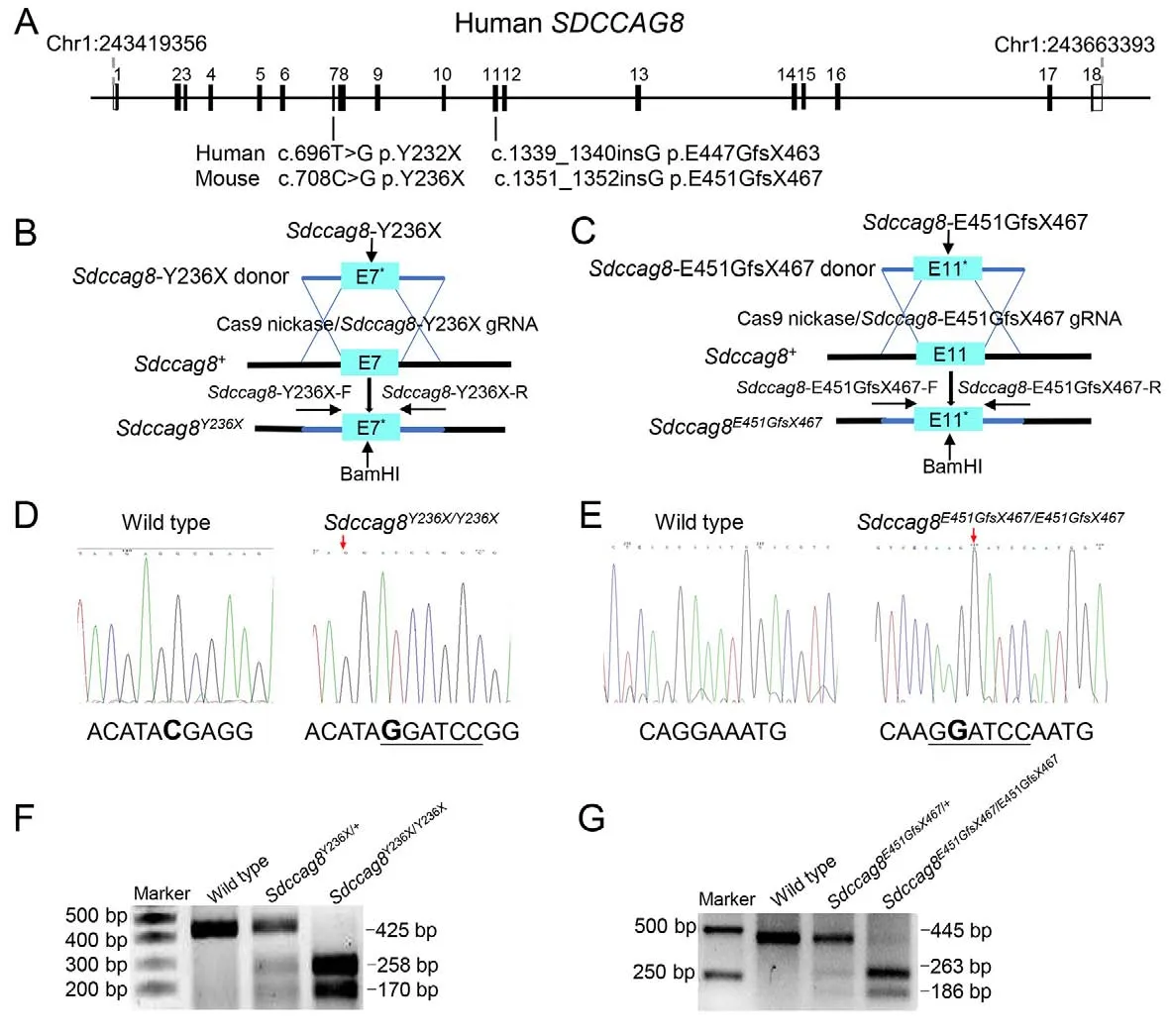
Figure 1 Generation of Sdccag8Y236X/Y236X and Sdccag8E451GfsX467/E451GfsX467 knock-in mice
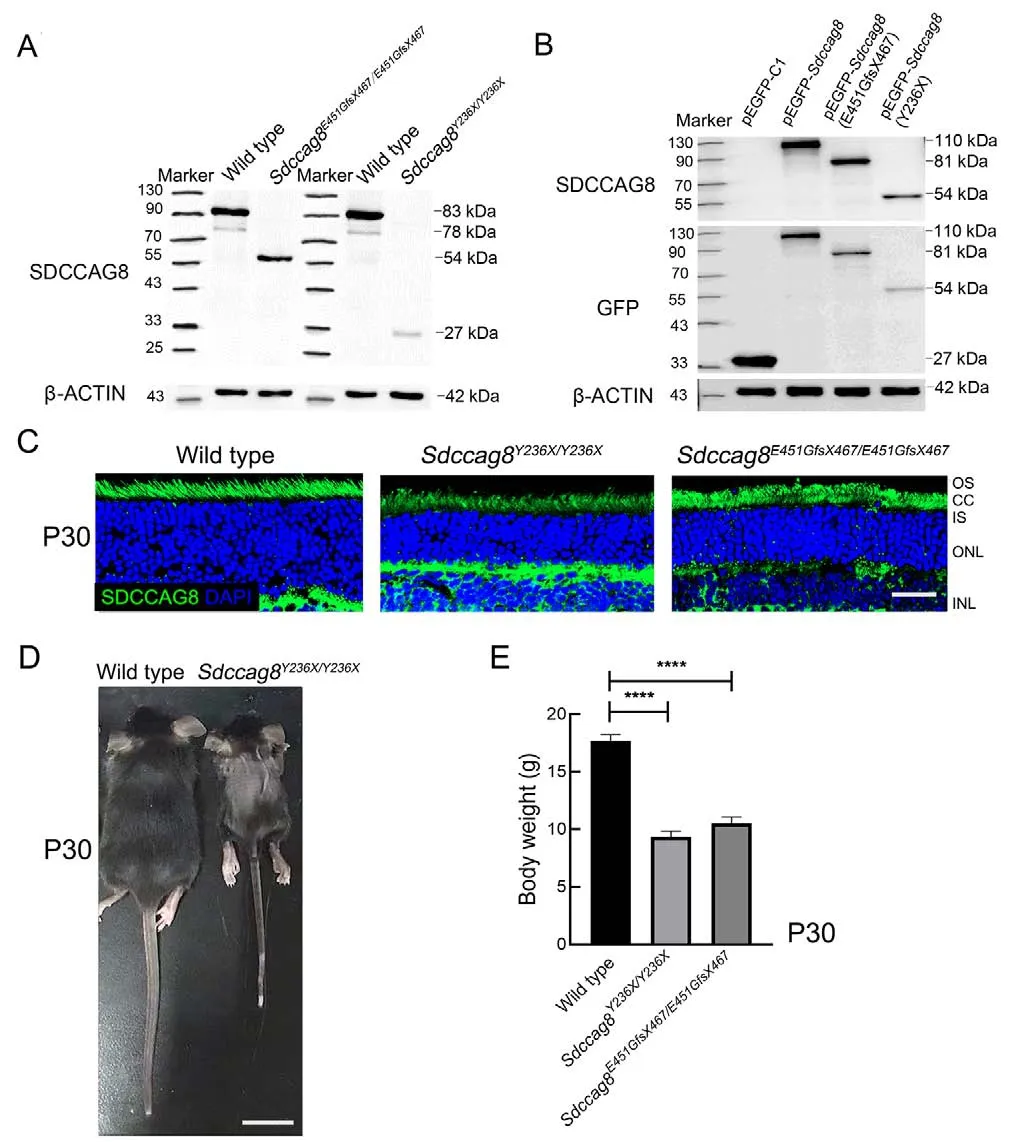
Figure 2 Sdccag8 knock-in mice carrying a hypomorphic allele with growth retardation
We next conducted scotopic and photopic ERGs on the knock-in mice to evaluate visual function of their rod and cone photoreceptors, respectively.For the scotopic ERGs, the awave amplitudes under 1.30 log cd·s/m2light stimuli significantly decreased by 53% inSdccag8Y236X/Y236Xmice(177.90±27.96 μV) and by 49% inSdccag8E451GfsX467/E451GfsX467mice (192.90±31.55 μV) at P30 compared to the wild-type controls (375.50±36.36 μV) (Figure 4A-C).The scotopic ERG responses recorded from both knock-in mice declined by 78%and 69% at P90 and by 93% and 85% by P180, respectively,(Figure 4A-C).For the photopic ERGs, the b-wave amplitudes under 1.30 log cd·s/m2light stimuli decreased by 33% inSdccag8E451GfsX467/E451GfsX467mice (116.40±20.58 μV) and by 38% inSdccag8Y236X/Y236Xmice (109.30±37.69 μV) at P30compared to the wild-type controls (174.10±20.93 μV)(Figure 4A, B, D).Progressively, the photopic ERG responses decreased by 71% inSdccag8Y236X/Y236Xmice (49.33±15.34 μV) and 60% inSdccag8E451GfsX467/E451GfsX467mice(66.75±12.36 μV) by P90, and further significantly declined in both knock-in mice (91% (15.33±6.01 μV) and 85%(26.67±7.17 μV), respectively) by P180 (Figure 4A, B, D),indicating that visual dysfunction caused by theSdccag8mutations was more severe inSdccag8Y236X/Y236Xmice than inSdccag8E451GfsX467/E451GfsX467mice, and was initiated earlier in rods than in cones.
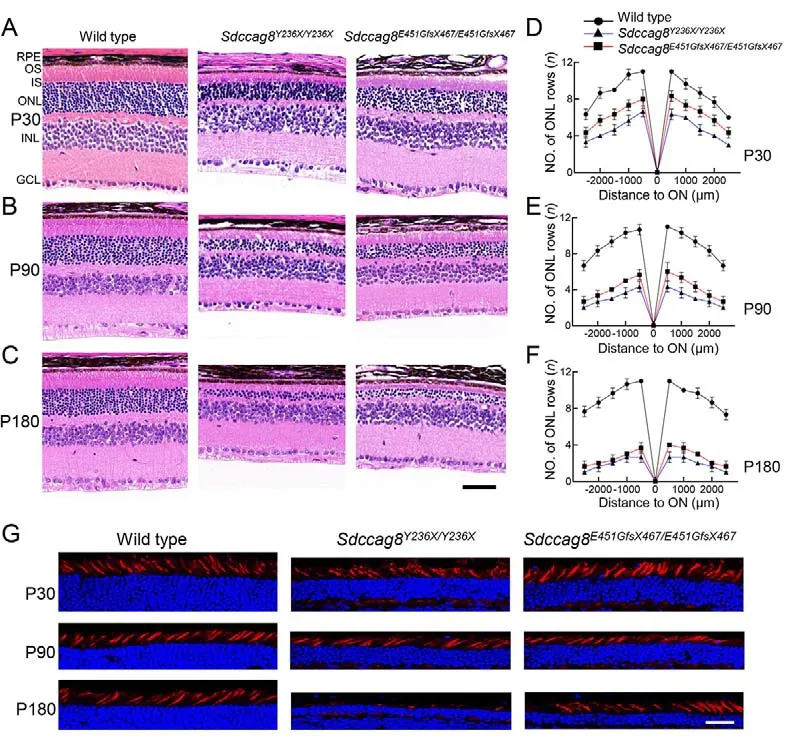
Figure 3 Retinal morphology of rod-cone photoreceptor degeneration in Sdccag8Y236X/Y236X and Sdccag8E451GfsX467/E451GfsX467 knock-in mice

Figure 4 Scotopic and photopic electroretinograms (ERG) of rod-cone photoreceptor degeneration in Sdccag8Y236X/Y236X and Sdccag8E451GfsX467/E451GfsX467 mice
Taken together, the retinal morphological and visual functional data of the knock-in mouse models demonstrated thatSdccag8mutations caused early-onset and rapidly progressive photoreceptor degeneration, which was more severe inSdccag8Y236X/Y236Xmice.In addition, rods degenerated earlier and faster than cones in both models,presenting as rod-cone degeneration.
Mislocalization of phototransduction proteins in Sdccag8 mutant mouse photoreceptors
Most retinal ciliopathy-causing mutations disrupt photoreceptor morphology and function through abrogation of phototransduction cascade components, trafficking defects of the photoreceptor OS proteins, and impaired cilium biogenesis or maintenance (Bujakowska et al., 2017; Reiter & Leroux,2017).Thus, to determine whether theSdccag8truncating mutations impaired ciliary protein trafficking in photoreceptors,we first examined the localization of several membrane and membrane-associated phototransduction proteins in the knock-in mice at P30 and P90 by immunohistochemical analysis.The transmembrane protein rhodopsin was observed in the photoreceptor OS inSdccag8E451GfsX467/E451GfsX467mice at P30 and P90, as in wild-type controls (Figure 5A, B).However, rhodopsin was mislocalized in the photoreceptor IS inSdccag8Y236X/Y236Xmice at P90, when the OS was significantly shortened (Figure 5A, B).Rhodopsin kinase(GRK1), a peripheral membrane protein involved in the phototransduction cascade, was localized in the shortened photoreceptor OS, with very little mistrafficking in the IS, ONL,and synaptic terminal inSdccag8Y236X/Y236XandSdccag8E451GfsX467/E451GfsX467retinas at P30 (Figure 5C).At P90, decreased mistrafficking of GRK1 was observed as the mutant photoreceptors were significantly decreased and their OSs were extensively shortened in both knock-in mice(Figure 5D).Likewise, PDE6b, another rod peripheral membrane protein, showed mistrafficking in the photoreceptors of the knock-in mice at both P30 and P90(Figure 5E, F).Subsequently, we examined the localization of two cone phototransduction proteins in the knock-in mouse retinas, namely membrane protein S-opsin and membraneassociated protein cone arrestin, which trafficked to the cone OS in the wild-type controls.We observed remarkable mislocalization of S-opsin and cone arrestin in the cone IS and synaptic terminal in the knock-in mice at P30 and P90(Figure 6A-D).Thus, our data revealed progressive mislocalization of OS-specific membrane proteins and membrane-associated proteins in the mutant photoreceptors,which was more obvious in the cones than in the rods.
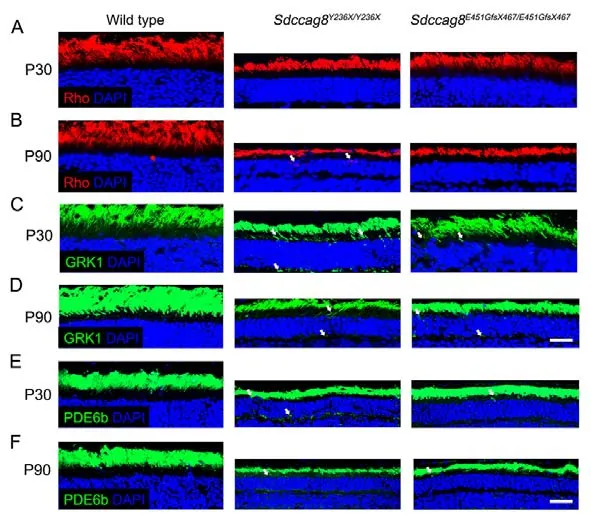
Figure 5 lmmunolocalization of rod phototransduction-related proteins in Sdccag8Y236X/Y236X and Sdccag8E451GfsX467/E451GfsX467 mouse retinas

Figure 6 lmmunolocalization of cone phototransduction-related proteins in Sdccag8Y236X/Y236X and Sdccag8E451GfsX467/E451GfsX467 mouse retinas
We subsequently tested whether protein mislocalization occurred before or after photoreceptor cell death by TUNEL staining of the knock-in mouse retinas at P21, before protein mislocalization occurred.TUNEL signals represent apoptosis,a predominant cell death mode of photoreceptor degeneration(Wright et al., 2010).We detected TUNEL signals in the ONL of bothSdccag8Y236X/Y236XandSdccag8E451GfsX467/E451GfsX467mice at P21, but not in wild-type controls (Figure 6E),revealing that OS protein mislocalization occurred after photoreceptor cell death in the knock-in mice, probably due to shortening of the photoreceptor OS, a sink of phototransduction proteins.
lmpaired photoreceptor cilia drive retinal degeneration in Sdccag8 knock-in mice
To test whether theSdccag8truncating mutations impaired photoreceptor cilia in the knock-in mice, we examined the photoreceptor ultrastructures of mutant mice at P60 by TEM,specifically focusing on the photoreceptor ciliary compartments, including BB, CC, and OS.The wild-type mice developed robust photoreceptor BB, CC, and OS compartments, with evenly stacked disk membranes(Figure 7A).In contrast, despite BB docking to photoreceptor apical membranes as usual, bothSdccag8Y236X/Y236XandSdccag8E451GfsX467/E451GfsX467mice exhibited shortened CC and disorganized OS compartments, with significantly deteriorated disk membranes (Figure 7A).These results revealed that the structure of the photoreceptor cilia in theSdccag8mutant mice was destroyed, suggesting that SDCCAG8 may function in photoreceptor cilium formation and/or maintenance.
We further studied whether theSdccag8truncating mutations affected global cilium formation and maintenance by examining primary cilium formation in MEFs derived from knock-in mice (Figure 7B).We assessed cilium occurrence and length of mutant MEFs by immunofluorescence staining with an antibody against the ciliary marker acetylated tubulin after serum starvation (Figure 7B-D).Primary cilium formationwas only found in 8% (8/100) ofSdccag8Y236X/Y236XMEFs and 39% (39/100) ofSdccag8E451GfsX467/E451GfsX467MEFs, in contrast to 80% (80/100) of wild-type control MEFs (Figure 7C).Additionally, the average lengths of cilia in theSdccag8Y236X/Y236XandSdccag8E451GfsX467/E451GfsX467MEFs were 0.87 μm and 2.10 μm, respectively, significantly shorter than that of the controls (Figure 7B, D).These results suggest that SDCCAG8 is essential for global cilium formation.
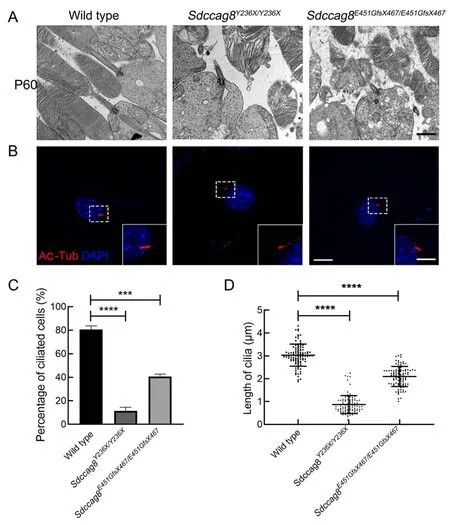
Figure 7 lmpaired cilia formation in photoreceptors and MEFs from Sdccag8Y236X/Y236X and Sdccag8E451GfsX467/E451GfsX467 mice
Our results revealed that impaired photoreceptor ciliary structure is a primary pathogenic force driving photoreceptor degeneration in the knock-in mice, with SDCCAG8 implicated to play a critical role in photoreceptor ciliary formation and maintenance, rather than ciliary protein trafficking.
Nephronophthisis and defective epithelial cilia in Sdccag8 knock-in mice
Patients with SDCCAG8-associated ciliopathies andSdccag8gt/gtgene-trap mice with loss ofSdccag8function exhibit NPHP, a renal cystic disease characterized by corticomedullary cysts, tubular basement membrane disruption, and tubulointerstitial nephropathy in renal histology(Stokman et al., 2016).Thus, we investigated whether the knock-in mice developed NPHP.
We observed kidney enlargement inSdccag8Y236X/Y236Xmice as early as P30, and the mutant kidneys became progressively larger at P90 and deformed at P180 (Figure 8A).Correspondingly, small cysts formed in the cortical region of theSdccag8Y236X/Y236Xmice at P30, as visualized by renal histological assay (Figure 8B).From P90 to P180, theSdccag8Y236X/Y236Xmouse kidneys progressively deteriorated,with renal cysts spreading beyond the cortical region to the corticomedullary junction, cortical cysts becoming enlarged,and renal parenchyma replaced by interstitial infiltrates(Figure 8B).However, different from early-onset NPHP inSdccag8Y236X/Y236Xmice, no shape change or cyst formation was detected in theSdccag8E451GfsX467/E451GfsX467mouse kidneys at P30 (Figure 8A, B).Relatively mild kidney enlargement and cyst formation in the kidney cortical region were observed in theSdccag8E451GfsX467/E451GfsX467mice at P90 to P180 (Figure 8A, B).To examine renal fibrosis in the knockin mice, we performed Masson trichrome staining of renal sections.Consistent with histology, theSdccag8Y236X/Y236Xmice showed rapidly progressive renal fibrosis, from mild fibrosis surrounding the dilated tubules at P30 and P90 to extensive collagen deposits distributed across the renal tissue at P180 (Figure 8C).Renal fibrosis was not detected in theSdccag8E451GfsX467/E451GfsX467mouse kidneys at P30 but was observed minimally at P90 and obviously at P180 (Figure 8C).Thus, our data demonstrated that NPHP occurred in both knock-in mouse models but with different severities andprogressed in concert with photoreceptor degeneration.

Figure 8 Nephronophthisis in Sdccag8Y236X/Y236X and Sdccag8E451GfsX467/E451GfsX467 mice accompanied by defective renal cilia
We next tested the uACR to investigate whether the knockin mice developed CKD, a typical clinical feature of NPHP and precursor to end-stage renal disease (ESRD).The uACR is a sensitive and specific surrogate marker for proteinuria,indicating progression of CKD (Hildebrandt et al., 2009).We collected 24 h urine samples from three P180 mice for each knock-in model and analyzed the uACR with an automatic biochemical analyzer.The 24 h uACRs inSdccag8Y236X/Y236X(132.5±7.0 μg/mg) andSdccag8E451GfsX467/E451GfsX467(59.4±7.0 μg/mg) mice were significantly elevated (3.3 and 1.5 times,respectively) compared to the wild-type controls (39.6±5.4 μg/mg).The uACR results indicated CKD progression in both knock-in mice by P180, with increased severity inSdccag8Y236X/Y236Xmice.
We next investigated whether theSdccag8mutations affected biogenesis of cilia in the mouse kidneys, similar to their effects on photoreceptors and MEFs.Renal cilia were examined by staining kidney sections with an antibody against the cilium marker acetylated tubulin.The number and length of renal epithelial cilia were significantly decreased in the distal convoluted tubules and cortical collecting ducts of theSdccag8E451GfsX467/E451GfsX467kidneys and were completely absent in theSdccag8Y236X/Y236Xkidneys (Figure 8D).
Taken together, our data demonstrate that the knock-in mice carryingSdccag8truncating mutations displayed NPHP and developed CKD, with earlier onset and increased severity inSdccag8Y236X/Y236Xmice.The ciliary defects detected in the kidney epithelial cells may be a driving force of renal cyst formation inSdccag8knock-in mice.
Hind limb preaxial polydactyly in Sdccag8 knock-in mice
Postaxial polydactyly is a major clinical feature of BBS in humans.Thus, we examined whether this phenotype was present in the knock-in mouse models.Interestingly, the mutant mice exhibited preaxial polydactyly of the hind limbs,with 100% (40/40) penetrance in theSdccag8Y236X/Y236Xmice and 95% (79/83) penetrance in theSdccag8E451GfsX467/E451GfsX467mice (Figure 9A, B).Bilateral polydactyly was predominant, accounting for 80% (32/40) of theSdccag8Y236X/Y236Xmice and 58% (48/83) of theSdccag8E451GfsX467/E451GfsX467mice (Figure 9B).Among the mice with unilateral polydactyly, 75% (6/8) of theSdccag8Y236X/Y236Xmice and 65% (20/31) of theSdccag8E451GfsX467/E451GfsX467mice presented with right-sided polydactyly, significantly more than left-sided polydactyly (Figure 9B).The development of polydactyly as a ciliopathy phenotype in the mouse models is consistent with previous findings in three gene-trap mouse models (Airik et al., 2016; Insolera et al., 2014; Weihbrecht et al., 2018), supporting the hypothesis that SDCCAG8 participates in digit development in mice.

Figure 9 Preaxial polydactyly phenotype in Sdccag8Y236X/Y236X and Sdccag8E451GfsX467/E451GfsX467 mice
DlSCUSSlON
In this study, two novelSdccag8knock-in mouse linesSdccag8Y236X/Y236XandSdccag8E451GfsX467/E451GfsX467, each carrying a distinct hypomorphic allele, were generated by CRISPR/Cas9-HDR.They faithfully recapitulated human SDCCAG8-associated BBS phenotypes with varied phenotypic age of onset and severity, which were directly proportional to the hypomorphic strength of theSdccag8mutations, Y236X and E451GfsX467.To the best of our knowledge, these knock-in mice were the first BBS mouse models to present with polydactyly.Impaired cilia were observed in the mutant photoreceptors, renal epithelial cells,and MEFs derived from the knock-in mouse embryos.Therefore, we propose that cilium defects are the primary driving force of SDCCAG8-associcated BBS in humans.
Due to a pivotal role of cilia in diverse cellular processes of embryonic and postnatal development (Reiter & Leroux,2017), most ciliopathy-associated mutations identified in humans are hypomorphic, includingSdccag8mutations,resulting in prenatal developmental defects, as well as clinical manifestations arising after birth.Thus, it is unsurprising that complete loss of a ciliopathy-associated gene is devastating to mice and humans (Norris & Grimes, 2012).Two out of threeSdccag8mutant mice previously generated by gene trap show embryonic lethality, with developmental defects in the central nervous system, limbs, and lungs, but without primary features of SDCCAG8-assciated ciliopathies, i.e., retinal and renal defects (Insolera et al., 2014; Weihbrecht et al., 2018).Therefore, conventional genetic modification approaches to generate null alleles may not be appropriate for modeling ciliopathy-associated diseases.The generation of hypomorphic alleles with a range of strengths for diseases that explicitly model human ciliopathies can be technically demanding (Rix et al., 2011).Among current genome engineering technologies, the CRISPR/Cas9 system is a programmable nuclease-based genome-editing technology that enables highly efficient and precise modification of avariety of eukaryotic and mammalian species (Banan, 2020;Hsu et al., 2014).Employing CRISPR/Cas9-HDR in this study,we successfully developed two mouse models carrying hypomorphic alleles ofSdccag8, which closely mimicked human BBS with phenotypic variation.
The mouseSdccag8mutationsSdccag8-Y236X andSdccag8-E451GfsX467 respectively correspond to BBS- and SLS-causing mutations in humans (Halbritter et al., 2013a;Otto et al., 2010).The corresponding knock-in mice generated in this study lacked the full-length SDCCAG8 protein and showed significantly decreased expression of the specific truncated proteins, in contrast to the null alleles described in three previously generatedSdccag8gene-trap mice (Airik et al., 2014; Insolera et al., 2014; Weihbrecht et al., 2018).The fewer truncated proteins could be attributed to nonsense mediated decay due to the inclusion of an early stop codon in theSdccag8mutant alleles.Previous genetic studies have suggested that only the full-length isoform SDCCAG8-a is relevant for the retinal-renal phenotype of SDCCAG8-associated ciliopathies, with specific expression in the photoreceptor CC and ISs of mouse retinas (Otto et al., 2010).The mutantsSDCCAG8-Y236X andSDCCAG8-E451GfsX467 caused different-sized truncations of the C-terminal CCD.Notably, based on immunochemical analysis of the knock-in mouse retinas, SDCCAG8-Y236X exhibited weaker fluorescence signals than SDCCAG8-E451GfsX467 in the mutant photoreceptor CC and ISs, suggesting that the size of the truncated CCD was directly proportional to the hypomorphic strength of the mutations.
Histological and ERG analysis of mouse retinas showed early-onset and rapidly progressive rod-cone photoreceptor degeneration in theSdccag8knock-in mice.Retinal degenerative changes, including photoreceptor ONL and OS shortening, were observed in theSdccag8Y236X/Y236Xmice as early as P30, and rapidly progressed to almost complete deterioration within 6 months.TheSdccag8E451GfsX467/E451GfsX467mice showed less severe morphological and functional changes in photoreceptor degeneration, progressing about one month later than theSdccag8Y236X/Y236Xmice.In addition,loss of cone photoreceptors was first observed in mutant retinas at P180, and ERG responses from cones decreased by ~20% in the knock-in mice at P30.In contrast, a 35%-50%loss of rods and 50% decrease in ERG responses from rods were observed in the knock-in mice even at P30.Thus, lateonset cone death and dysfunction revealed rod-cone photoreceptor degeneration in SDCCAG8-associated retinal ciliopathies.
In addition, early- and late-onset NPHP developed in the two knock-in mouse models, respectively, with formation of renal cysts inSdccag8Y236X/Y236Xmice as early as P30, but at P90 inSdccag8E451GfsX467/E451GfsX467mice.However, among the three previously generatedSdccag8gene-trap mouse models,onlySdccag8gt/gtmice exhibited late-onset NPHP, with embryonic lethality reported in the other mice (Airik et al.,2014; Insolera et al., 2014; Weihbrecht et al., 2018).NPHP is a cystic renal disease that constitutes the most frequent genetic cause of ESRD in children and young adults, and is characterized by corticomedullary cysts, tubular basement membrane disruption, and tubulointerstitial nephropathy in renal histology (Stokman et al., 2016).TheSdccag8knock-in mice displayed moderately enlarged and/or deformed kidneys,not unlike infantile NPHP in humans (Airik et al., 2014; Braun& Hildebrandt, 2017; Kang et al., 2016; Otto et al., 2010).Previous mouse models of cystic renal disease have shown renal cysts in the cortical region during the early stages, with spread to the corticomedullary junction and interstitial infiltrates at the end stage (Atala et al., 1993; Attanasio et al.,2007).Similarly, our mouse models demonstrated renal fibrosis surrounding dilated tubules during early disease, and later widespread deterioration of renal tissue.Here, mouse renal function was evaluated by 24 h uACR testing, which indicated excessive proteinuria inSdccag8Y236X/Y236Xmice at P180, consistent with ESRD.The different severities of NPHP in the two knock-in mouse models further demonstrated the different hypomorphic strengths of theSdccag8-Y236X andSdccag8-E451GfsX467 mutant alleles.
The three previously reportedSdccag8gene-trap mouse lines exhibit preaxial polydactyly penetrance of 65%-100%(Airik et al., 2016; Insolera et al., 2014; Weihbrecht et al.,2018), similar to the two knock-in mouse lines at 95% and 100%, respectively.As an embryonically developed phenotype, polydactyly is a primary feature of BBS and other ciliopathies, such as MKS, JBTS, orofaciodigital syndrome(OFD), and McKusick-Kaufman syndrome (MKKS), but not SLS (Zaghloul & Katsanis, 2009).About 79% of BBS patients present with postaxial polydactyly, but the polydactyly phenotype is absent in human SDCCAG8-assciated ciliopathies (Beales et al., 1999; Forsythe & Beales, 2013;Otto et al., 2010; Schaefer et al., 2011).To the best of our knowledge, theSdccag8-associated mutant mice, including the gene-trap and knock-in mice, are the first BBS mouse models characterized by digital malformation.In addition,instead of presenting postaxial polydactyly on any limb, as seen in BBS patients, theSdccag8-associated mouse models exhibited preaxial polydactyly on hind limbs only.Many ciliary gene mutations causing polydactyly are attributed to disruption of the Sonic Hedgehog (Shh) signaling pathway, which requires the presence of intact primary cilia for activation(Zaghloul & Katsanis, 2009).The presence of polydactyly in the knock-in mice suggests that SDCCAG8 is necessary for cilium biogenesis and/or maintenance of the developing limb bud in mice, but not in humans.Furthermore, this suggests species-specific differences in Shh-dependent determination of digit number and identity.Therefore, further studies are needed to improve our understanding of the underlying pathogenesis of polydactyly in human BBS.
The twoSdccag8knock-in mouse lines were characterized as BBS animal models, displaying four out of the six BBS primary manifestations, including rod-cone dystrophy, cystic renal disorder, polydactyly, and male infertility, as well as secondary developmental delay.Phenotypic severity and age of onset were dependent on the hypomorphic strength of theSdccag8mutations, which was directly correlated to truncation size and expression level of the mutant alleles.
We also explored the pathogenesis of SDCCAG8 mutationcausing retinal ciliopathies by dissecting OS protein trafficking and ciliary structure of the mutant photoreceptors in theSdccag8knock- in mice.We discovered rodphototransduction-related proteins, including rhodopsin,GRK1, and PDE6, localized not only in the OS, but also in the photoreceptor IS, ONL, and/or synaptic terminals of the knockin mice at P90, when rod photoreceptors were dying and their OSs were significantly shortened.In addition, cone phototransduction-related proteins S-opsin and cone arrestin were significantly mislocalized in the IS, ONL, and synaptic terminals at P30, which was probably due to differences in OS biogenesis or protein trafficking between cones and rods(Anderson et al., 1978; Eckmiller, 1987).However, based on TUNEL staining, we found that photoreceptor cell death occurred earlier than OS protein mislocalization in the mutant photoreceptors, suggesting thatSdccag8mutation-causing protein mislocalization occurred after photoreceptor cell death.Impaired ciliary structures, including shortened CC and disrupted OS, were detected in the knock-in mice by TEM analysis of retinas.Impaired renal epithelial cilia were observed in the distal convoluted tubules and cortical collecting ducts of the mutant kidneys.In addition,Sdccag8mutation-causing global cilium defects were verified in MEFs derived from the knock-in mice.Thus, our data demonstrated that cilium defects were a primary force driving BBS phenotypes in the knock-in mice, suggesting that SDCCAG8 plays an essential role in biogenesis and maintenance of cilia in multiple systems during embryonic development and after birth.Notably, we found that SDCCAG8 is a ciliary protein involved in retinal ciliopathies and plays a key role in cilium assembly and/or maintenance.Further analysis of its role should provide insights into ciliary function and increase our understanding of the signaling, physiological, and developmental functions of cilia.
COMPETlNG lNTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRlBUTlONS
L.J.and Z.L.Y.designed the study and wrote and revised the manuscript.Z.L.R.performed the experiments, analyzed the data, and drafted the manuscript.H.B.Z.and L.L.helped analyze the data and reviewed and edited the manuscript.All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We thank Shu-Jin Li, Mu Yang, and Shan-Shan Zhang for assistance in the immunochemical experiments and Jia-Liang Yang and Zhi-Lin Jiang for assistance in the ERG experiment.We also thank Lin Wang for help in breeding mice and Tian-Ge Song and Ke-Cheng Li for help in obtaining tail clips from mice.
杂志排行

Zoological Research的其它文章
- Adult hippocampal neurogenesis and its impairment in Alzheimer’s disease
- Discovery of a wild, genetically pure Chinese giant salamander creates new conservation opportunities
- Combinational benefit of antihistamines and remdesivir for reducing SARS-CoV-2 replication and alleviating inflammation-induced lung injury in mice
- Scan of the endogenous retrovirus sequences across the swine genome and survey of their copy number variation and sequence diversity among various Chinese and Western pig breeds
- Novel astrovirus and paramyxovirus in Mongolian gerbils (Meriones unguiculatus) from China
- A new species of the genus Typhlomys Milne-Edwards,1877 (Rodentia: Platacanthomyidae) from Chongqing,China